1강: 표현형 기반 임상 데이터 기초 통계 분석
BBP WGS 분석 튜토리얼
학습 목표
- 표현형 기반 임상 데이터 구조 이해 - 1000 Genomes Project의 샘플/표현형 데이터 구조를 파악
- 임상 정보 통계 및 연관성 분석 - 인구집단별, 성별 등 임상 변수의 기초 통계 및 연관성 분석
- 유전변이(VCF)-임상 표현형 통합 분석을 위한 전처리 - VCF 데이터와 임상 데이터를 결합하기 위한 데이터 정제
사용 데이터
데이터 루트: /tier4/DSC/jheepark/bbp-wgs-data/
| 데이터 | 경로 | 설명 |
|---|---|---|
| 샘플 패널 정보 | phenotype/integrated_call_samples_v3.20130502.ALL.panel | 2,504 샘플의 population / super_population / gender |
| 샘플 상세 정보 | phenotype/20130606_sample_info.xlsx | 샘플별 상세 메타데이터 (시퀀싱 플랫폼, 커버리지 등) |
| Pedigree (가계도) | phenotype/20130606_g1k.ped | 가족 관계, 성별, 표현형 정보 (PED 형식) |
| 1000 Genomes chr22 VCF | individual/ALL.chr22.phase3_shapeit2_mvncall_integrated_v5b.20130502.genotypes.vcf.gz | VCF 샘플 목록 매칭 검증용 |
산출물 (본 강의에서 생성)
| 파일 | 경로 | 설명 |
|---|---|---|
| 통합 샘플 정보 | phenotype/integrated_sample_info.tsv | Panel + VCF 샘플 순서 병합 (2–4강에서 재사용) |
| EAS 서브셋 | phenotype/eas_sample_info.tsv | 동아시아(EAS) 504 샘플 |
| Unrelated 샘플 목록 | phenotype/unrelated_samples.txt | 부모 정보 없는 unrelated 샘플 ID (4강 GWAS용) |
사용 도구
- Python: pandas, numpy, matplotlib, seaborn, scipy
- bcftools (VCF 샘플 목록 확인용)
0. 환경 설정
python
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
from scipy import stats
from matplotlib import font_manager, rc
import warnings
warnings.filterwarnings('ignore')
# 시각화 설정 (seaborn 스타일을 먼저 설정해야 한글 폰트가 덮어씌워지지 않음)
sns.set_style('whitegrid')
sns.set_palette('Set2')
# 한글 폰트 설정 (seaborn 설정 이후에 적용)
font_path = '/usr/share/fonts/truetype/nanum/NanumGothic.ttf'
font_manager.fontManager.addfont(font_path)
rc('font', family='NanumGothic')
plt.rcParams['axes.unicode_minus'] = False # 마이너스 기호 깨짐 방지
plt.rcParams['figure.figsize'] = (12, 6)
plt.rcParams['font.size'] = 12
# 데이터 경로 설정
DATA_DIR = '/tier4/DSC/jheepark/bbp-wgs-data'
PHENOTYPE_DIR = f'{DATA_DIR}/phenotype'
INDIVIDUAL_DIR = f'{DATA_DIR}/individual'
print('환경 설정 완료!')text
환경 설정 완료!1. 표현형 기반 임상 데이터 구조 이해
1000 Genomes Project는 전 세계 26개 인구집단에서 2,504명의 개인을 대상으로 whole genome sequencing을 수행한 대규모 유전체 프로젝트입니다.
1.1 샘플 패널 데이터 로드
.panel 파일은 각 샘플의 인구집단(population), 대륙 그룹(super population), 성별(gender) 등 기본 정보를 담고 있습니다.
python
# 샘플 패널 정보 로드
panel = pd.read_csv(
f'{PHENOTYPE_DIR}/integrated_call_samples_v3.20130502.ALL.panel',
sep='\t'
)
print(f'전체 샘플 수: {len(panel)}')
print(f'컬럼: {list(panel.columns)}')
print()
panel.head(10)text
전체 샘플 수: 2504
컬럼: ['sample', 'pop', 'super_pop', 'gender', 'Unnamed: 4', 'Unnamed: 5']| sample | pop | super_pop | gender | Unnamed: 4 | Unnamed: 5 | |
|---|---|---|---|---|---|---|
| 0 | HG00096 | GBR | EUR | male | NaN | NaN |
| 1 | HG00097 | GBR | EUR | female | NaN | NaN |
| 2 | HG00099 | GBR | EUR | female | NaN | NaN |
| 3 | HG00100 | GBR | EUR | female | NaN | NaN |
| 4 | HG00101 | GBR | EUR | male | NaN | NaN |
| 5 | HG00102 | GBR | EUR | female | NaN | NaN |
| 6 | HG00103 | GBR | EUR | male | NaN | NaN |
| 7 | HG00105 | GBR | EUR | male | NaN | NaN |
| 8 | HG00106 | GBR | EUR | female | NaN | NaN |
| 9 | HG00107 | GBR | EUR | male | NaN | NaN |
python
# 데이터 기본 정보 확인
print('=== 데이터 타입 ====')
print(panel.dtypes)
print()
print('=== 결측치 확인 ====')
print(panel.isnull().sum())
print()
print('=== 기초 통계 ====')
panel.describe(include='all')text
=== 데이터 타입 ====
sample object
pop object
super_pop object
gender object
Unnamed: 4 float64
Unnamed: 5 float64
dtype: object
=== 결측치 확인 ====
sample 0
pop 0
super_pop 0
gender 0
Unnamed: 4 2504
Unnamed: 5 2504
dtype: int64
=== 기초 통계 ====| sample | pop | super_pop | gender | Unnamed: 4 | Unnamed: 5 | |
|---|---|---|---|---|---|---|
| count | 2504 | 2504 | 2504 | 2504 | 0.0 | 0.0 |
| unique | 2504 | 26 | 5 | 2 | NaN | NaN |
| top | NA21144 | GWD | AFR | female | NaN | NaN |
| freq | 1 | 113 | 661 | 1271 | NaN | NaN |
| mean | NaN | NaN | NaN | NaN | NaN | NaN |
| std | NaN | NaN | NaN | NaN | NaN | NaN |
| min | NaN | NaN | NaN | NaN | NaN | NaN |
| 25% | NaN | NaN | NaN | NaN | NaN | NaN |
| 50% | NaN | NaN | NaN | NaN | NaN | NaN |
| 75% | NaN | NaN | NaN | NaN | NaN | NaN |
| max | NaN | NaN | NaN | NaN | NaN | NaN |
1.2 Super Population (대륙 그룹) 이해
1000 Genomes Project는 5개의 대륙 그룹(Super Population)으로 분류됩니다:
- AFR: African (아프리카)
- AMR: Ad Mixed American (아메리카 혼혈)
- EAS: East Asian (동아시아)
- EUR: European (유럽)
- SAS: South Asian (남아시아)
python
# Super Population별 샘플 수
superpop_counts = panel['super_pop'].value_counts()
print('=== Super Population별 샘플 수 ===')
print(superpop_counts)
print()
# Population별 샘플 수
pop_counts = panel['pop'].value_counts()
print('=== Population별 샘플 수 ===')
print(pop_counts)text
=== Super Population별 샘플 수 ===
super_pop
AFR 661
EAS 504
EUR 503
SAS 489
AMR 347
Name: count, dtype: int64
=== Population별 샘플 수 ===
pop
GWD 113
YRI 108
IBS 107
TSI 107
CHS 105
PUR 104
JPT 104
CHB 103
GIH 103
ITU 102
STU 102
ESN 99
CEU 99
LWK 99
FIN 99
KHV 99
PJL 96
ACB 96
CLM 94
CDX 93
GBR 91
BEB 86
PEL 85
MSL 85
MXL 64
ASW 61
Name: count, dtype: int64python
# Super Population 분포 시각화
fig, axes = plt.subplots(1, 2, figsize=(16, 6))
# 바 차트
superpop_colors = {'AFR': '#e74c3c', 'AMR': '#f39c12', 'EAS': '#2ecc71', 'EUR': '#3498db', 'SAS': '#9b59b6'}
colors = [superpop_colors[sp] for sp in superpop_counts.index]
axes[0].bar(superpop_counts.index, superpop_counts.values, color=colors, edgecolor='black', linewidth=0.5)
axes[0].set_title('Super Population별 샘플 수', fontsize=14, fontweight='bold')
axes[0].set_xlabel('Super Population')
axes[0].set_ylabel('샘플 수')
for i, (idx, val) in enumerate(superpop_counts.items()):
axes[0].text(i, val + 5, str(val), ha='center', fontweight='bold')
# 파이 차트
axes[1].pie(superpop_counts.values, labels=superpop_counts.index, autopct='%1.1f%%',
colors=colors, startangle=90, textprops={'fontsize': 12})
axes[1].set_title('Super Population 비율', fontsize=14, fontweight='bold')
plt.tight_layout()
plt.show()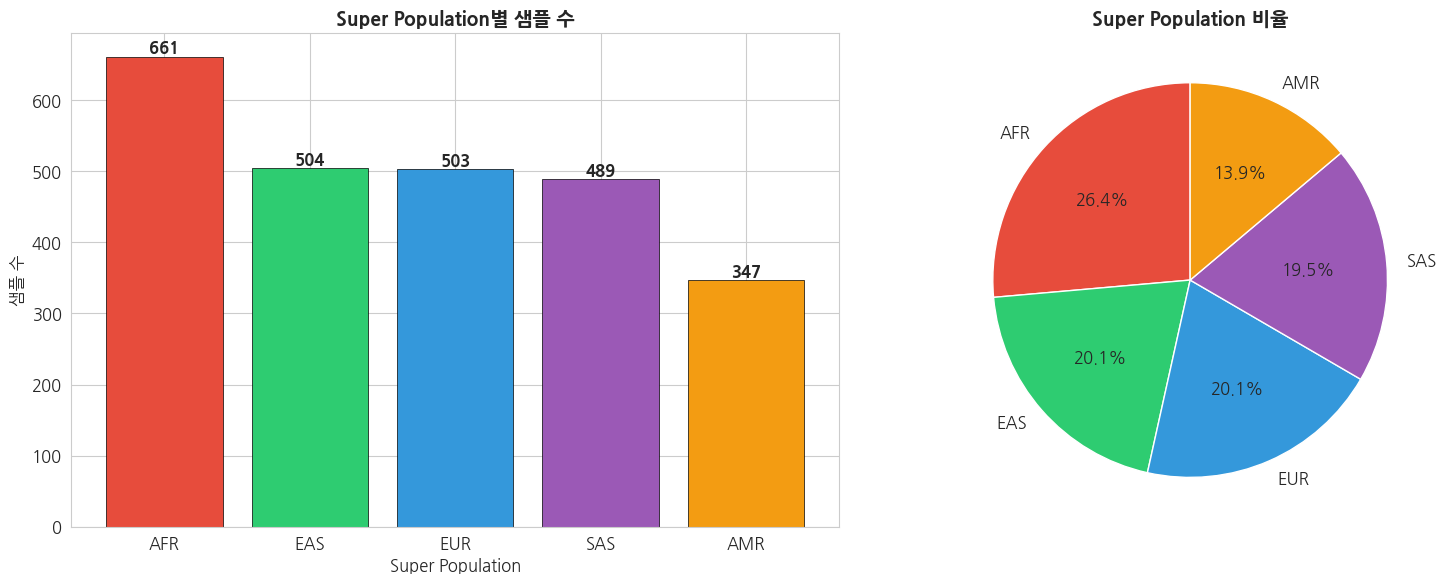
1.3 세부 Population 분포
python
# Population별 Super Population 매핑 테이블 생성
pop_superpop = panel.groupby(['super_pop', 'pop']).size().reset_index(name='count')
pop_superpop = pop_superpop.sort_values(['super_pop', 'count'], ascending=[True, False])
# 26개 Population 시각화
fig, ax = plt.subplots(figsize=(16, 8))
# Super Population별 색상 지정
bar_colors = [superpop_colors[row['super_pop']] for _, row in pop_superpop.iterrows()]
bars = ax.bar(range(len(pop_superpop)), pop_superpop['count'].values, color=bar_colors, edgecolor='black', linewidth=0.5)
ax.set_xticks(range(len(pop_superpop)))
ax.set_xticklabels(pop_superpop['pop'].values, rotation=45, ha='right')
ax.set_title('26개 Population별 샘플 수', fontsize=14, fontweight='bold')
ax.set_xlabel('Population')
ax.set_ylabel('샘플 수')
# 범례
from matplotlib.patches import Patch
legend_elements = [Patch(facecolor=superpop_colors[sp], edgecolor='black', label=sp) for sp in ['AFR', 'AMR', 'EAS', 'EUR', 'SAS']]
ax.legend(handles=legend_elements, title='Super Population', loc='upper right')
plt.tight_layout()
plt.show()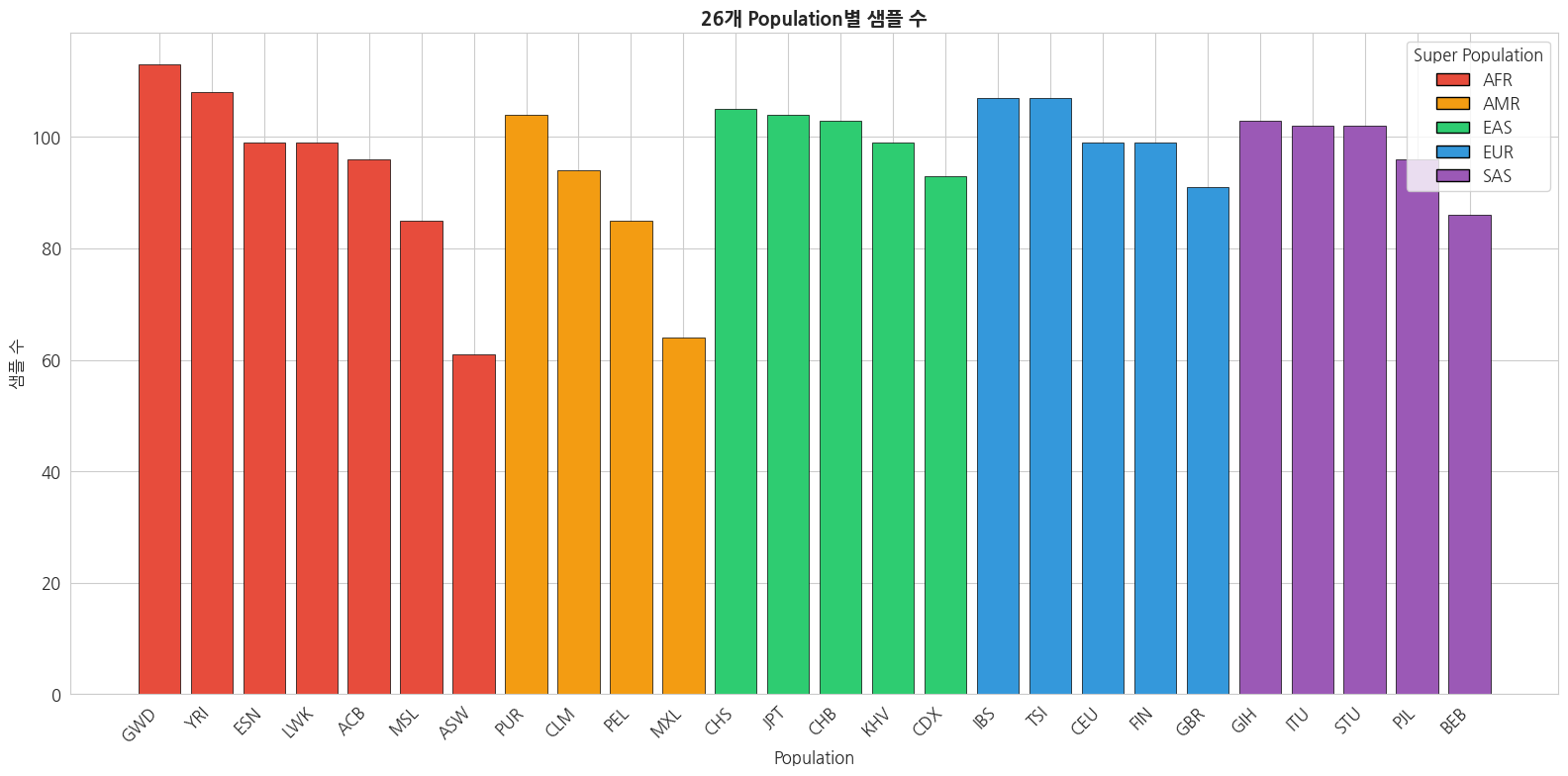
1.4 Pedigree (가계도) 데이터 로드
PED 파일은 가계 관계, 성별, 표현형 등의 정보를 포함합니다. PLINK에서 사용하는 표준 형식이기도 합니다.
python
# Pedigree 데이터 로드
ped_columns = [
'Family_ID', 'Individual_ID', 'Paternal_ID', 'Maternal_ID',
'Gender', 'Phenotype',
'Population', 'Relationship', 'Siblings', 'Second_Order',
'Third_Order', 'Other_Comments'
]
ped = pd.read_csv(
f'{PHENOTYPE_DIR}/20130606_g1k.ped',
sep='\t',
header=0
)
print(f'Pedigree 데이터 샘플 수: {len(ped)}')
print(f'컬럼: {list(ped.columns)}')
print()
ped.head(10)text
Pedigree 데이터 샘플 수: 3501
컬럼: ['Family ID', 'Individual ID', 'Paternal ID', 'Maternal ID', 'Gender', 'Phenotype', 'Population', 'Relationship', 'Siblings', 'Second Order', 'Third Order', 'Other Comments']| Family ID | Individual ID | Paternal ID | Maternal ID | Gender | Phenotype | Population | Relationship | Siblings | Second Order | Third Order | Other Comments | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | BB01 | HG01879 | 0 | 0 | 1 | 0 | ACB | father | 0 | 0 | 0 | 0 |
| 1 | BB01 | HG01880 | 0 | 0 | 2 | 0 | ACB | mother | 0 | 0 | 0 | 0 |
| 2 | BB01 | HG01881 | HG01879 | HG01880 | 2 | 0 | ACB | child | 0 | 0 | 0 | 0 |
| 3 | BB02 | HG01882 | 0 | 0 | 1 | 0 | ACB | father | 0 | 0 | 0 | 0 |
| 4 | BB02 | HG01883 | 0 | 0 | 2 | 0 | ACB | mother | 0 | 0 | 0 | 0 |
| 5 | BB02 | HG01888 | HG01882 | HG01883 | 1 | 0 | ACB | child | 0 | 0 | 0 | 0 |
| 6 | BB03 | HG01884 | HG01885 | HG01956 | 2 | 0 | ACB | child | 0 | 0 | 0 | 0 |
| 7 | BB03 | HG01885 | 0 | 0 | 1 | 0 | ACB | father | 0 | 0 | 0 | 0 |
| 8 | BB03 | HG01956 | 0 | 0 | 2 | 0 | ACB | mother | 0 | 0 | 0 | 0 |
| 9 | BB04 | HG01886 | 0 | 0 | 2 | 0 | ACB | mother | 0 | 0 | 0 | 0 |
python
# PED 파일 주요 필드 설명
print('=== PED 파일 주요 필드 ===')
print('Family_ID : 가족 ID')
print('Individual_ID : 개인 ID (VCF 샘플명과 매칭)')
print('Paternal_ID : 아버지 ID (0 = 정보없음)')
print('Maternal_ID : 어머니 ID (0 = 정보없음)')
print('Gender : 성별 (1=Male, 2=Female)')
print('Phenotype : 표현형 (0=Missing, 1=Unaffected, 2=Affected)')
print()
print('=== Gender 분포 ===')
gender_col = [c for c in ped.columns if 'ender' in c.lower()][0]
print(ped[gender_col].value_counts())
print()
print('=== Relationship 분포 ===')
rel_col = [c for c in ped.columns if 'elation' in c.lower()][0]
print(ped[rel_col].value_counts())text
=== PED 파일 주요 필드 ===
Family_ID : 가족 ID
Individual_ID : 개인 ID (VCF 샘플명과 매칭)
Paternal_ID : 아버지 ID (0 = 정보없음)
Maternal_ID : 어머니 ID (0 = 정보없음)
Gender : 성별 (1=Male, 2=Female)
Phenotype : 표현형 (0=Missing, 1=Unaffected, 2=Affected)
=== Gender 분포 ===
Gender
2 1761
1 1740
Name: count, dtype: int64
=== Relationship 분포 ===
Relationship
unrel 1003
mother 831
father 816
child 693
mat grandmother 31
mat grandfather 29
pat grandmother 28
pat grandfather 28
unrels 14
Child2 7
not father 3
daughter 1
father; child 1
pat grandfather; father 1
mother; child 1
pat grandmother; mother 1
mat grandfather; father 1
Child 1
mat grandmother; mother 1
wife of child 1
paternal father 1
paternal brother 1
child of 19672 1
child of 19740 1
child of 19752&3 1
child of 19764 1
husband of Child 1
maternal grandmother 1
paternal grandmother 1
Name: count, dtype: int641.5 샘플 상세 정보 (Excel)
python
# 샘플 상세 정보 로드
sample_info = pd.read_excel(
f'{PHENOTYPE_DIR}/20130606_sample_info.xlsx',
engine='openpyxl'
)
print(f'샘플 상세 정보 수: {len(sample_info)}')
print(f'컬럼 수: {len(sample_info.columns)}')
print(f'\n컬럼 목록:')
for i, col in enumerate(sample_info.columns, 1):
print(f' {i:2d}. {col}')
print()
sample_info.head()text
샘플 상세 정보 수: 3500
컬럼 수: 15
컬럼 목록:
1. Sample
2. Family ID
3. Population
4. Population Description
5. Gender
6. Relationship
7. Unexpected Parent/Child
8. Non Paternity
9. Siblings
10. Grandparents
11. Avuncular
12. Half Siblings
13. Unknown Second Order
14. Third Order
15. Other Comments| Sample | Family ID | Population | Population Description | Gender | Relationship | Unexpected Parent/Child | Non Paternity | Siblings | Grandparents | Avuncular | Half Siblings | Unknown Second Order | Third Order | Other Comments | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | HG00096 | HG00096 | GBR | British in England and Scotland | male | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| 1 | HG00097 | HG00097 | GBR | British in England and Scotland | female | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| 2 | HG00098 | HG00098 | GBR | British in England and Scotland | male | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| 3 | HG00099 | HG00099 | GBR | British in England and Scotland | female | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| 4 | HG00100 | HG00100 | GBR | British in England and Scotland | female | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
2. 임상 정보 통계 및 연관성 분석
2.1 데이터 통합
Panel 데이터와 Pedigree 데이터를 결합하여 종합적인 분석 데이터셋을 구축합니다.
python
# Panel 데이터와 Pedigree 데이터 병합
# panel의 'sample' 컬럼과 ped의 'Individual ID' 컬럼으로 매칭
ped_id_col = [c for c in ped.columns if 'ndividual' in c][0]
merged = panel.merge(
ped,
left_on='sample',
right_on=ped_id_col,
how='left'
)
print(f'병합 전 Panel 샘플 수: {len(panel)}')
print(f'병합 전 Pedigree 샘플 수: {len(ped)}')
print(f'병합 후 샘플 수: {len(merged)}')
print(f'\n병합 데이터 컬럼: {list(merged.columns)}')text
병합 전 Panel 샘플 수: 2504
병합 전 Pedigree 샘플 수: 3501
병합 후 샘플 수: 2504
병합 데이터 컬럼: ['sample', 'pop', 'super_pop', 'gender', 'Unnamed: 4', 'Unnamed: 5', 'Family ID', 'Individual ID', 'Paternal ID', 'Maternal ID', 'Gender', 'Phenotype', 'Population', 'Relationship', 'Siblings', 'Second Order', 'Third Order', 'Other Comments']2.2 성별(Gender) 분포 분석
python
# 성별 분포 - 전체 및 Super Population별
fig, axes = plt.subplots(1, 3, figsize=(18, 5))
# 전체 성별 분포
gender_counts = panel['gender'].value_counts()
axes[0].bar(gender_counts.index, gender_counts.values, color=['#3498db', '#e74c3c'], edgecolor='black')
axes[0].set_title('전체 성별 분포', fontsize=14, fontweight='bold')
axes[0].set_ylabel('샘플 수')
for i, (idx, val) in enumerate(gender_counts.items()):
axes[0].text(i, val + 5, str(val), ha='center', fontweight='bold')
# Super Population별 성별 분포
gender_by_superpop = panel.groupby(['super_pop', 'gender']).size().unstack(fill_value=0)
gender_by_superpop.plot(kind='bar', ax=axes[1], color=['#e74c3c', '#3498db'], edgecolor='black', linewidth=0.5)
axes[1].set_title('Super Population별 성별 분포', fontsize=14, fontweight='bold')
axes[1].set_xlabel('Super Population')
axes[1].set_ylabel('샘플 수')
axes[1].legend(title='Gender')
axes[1].tick_params(axis='x', rotation=0)
# 성별 비율 (stacked)
gender_ratio = gender_by_superpop.div(gender_by_superpop.sum(axis=1), axis=0)
gender_ratio.plot(kind='bar', stacked=True, ax=axes[2], color=['#e74c3c', '#3498db'], edgecolor='black', linewidth=0.5)
axes[2].set_title('Super Population별 성별 비율', fontsize=14, fontweight='bold')
axes[2].set_xlabel('Super Population')
axes[2].set_ylabel('비율')
axes[2].legend(title='Gender')
axes[2].tick_params(axis='x', rotation=0)
axes[2].axhline(y=0.5, color='black', linestyle='--', alpha=0.5)
plt.tight_layout()
plt.show()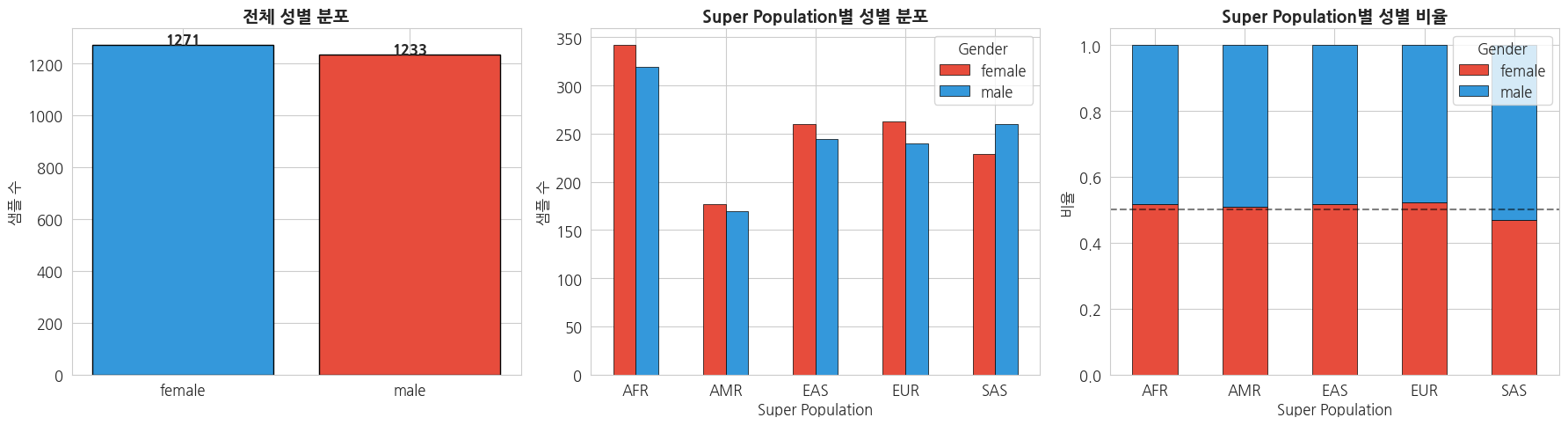
2.3 카이제곱 검정 (Chi-square Test)
Super Population과 성별 간에 통계적으로 유의미한 연관성이 있는지 확인합니다.
python
# Super Population × Gender 교차표
contingency_table = pd.crosstab(panel['super_pop'], panel['gender'])
print('=== 교차표 (Contingency Table) ===')
print(contingency_table)
print()
# 카이제곱 검정
chi2, p_value, dof, expected = stats.chi2_contingency(contingency_table)
print('=== 카이제곱 검정 결과 ===')
print(f'Chi-square statistic: {chi2:.4f}')
print(f'P-value: {p_value:.4f}')
print(f'Degrees of freedom: {dof}')
print()
if p_value < 0.05:
print('→ p < 0.05: Super Population과 성별 간에 통계적으로 유의미한 연관성이 있습니다.')
else:
print('→ p >= 0.05: Super Population과 성별 간에 통계적으로 유의미한 연관성이 없습니다.')
print(' (각 Super Population에서 남녀 비율이 비슷하게 설계되었음을 의미합니다.)')
print()
print('=== 기대빈도 (Expected Frequencies) ===')
expected_df = pd.DataFrame(expected, index=contingency_table.index, columns=contingency_table.columns)
print(expected_df.round(1))text
=== 교차표 (Contingency Table) ===
gender female male
super_pop
AFR 342 319
AMR 177 170
EAS 260 244
EUR 263 240
SAS 229 260
=== 카이제곱 검정 결과 ===
Chi-square statistic: 3.8906
P-value: 0.4210
Degrees of freedom: 4
→ p >= 0.05: Super Population과 성별 간에 통계적으로 유의미한 연관성이 없습니다.
(각 Super Population에서 남녀 비율이 비슷하게 설계되었음을 의미합니다.)
=== 기대빈도 (Expected Frequencies) ===
gender female male
super_pop
AFR 335.5 325.5
AMR 176.1 170.9
EAS 255.8 248.2
EUR 255.3 247.7
SAS 248.2 240.82.4 Population별 샘플 크기 분석
python
# Population별 성별 구성 히트맵
pop_gender = pd.crosstab(panel['pop'], panel['gender'])
pop_gender['total'] = pop_gender.sum(axis=1)
pop_gender = pop_gender.sort_values('total', ascending=False)
# Super Population 정보 추가
pop_to_superpop = panel.drop_duplicates('pop').set_index('pop')['super_pop']
fig, ax = plt.subplots(figsize=(10, 10))
sns.heatmap(
pop_gender[['female', 'male']],
annot=True, fmt='d', cmap='YlOrRd',
linewidths=0.5, ax=ax
)
ax.set_title('Population별 성별 분포 히트맵', fontsize=14, fontweight='bold')
ax.set_ylabel('Population')
ax.set_xlabel('Gender')
plt.tight_layout()
plt.show()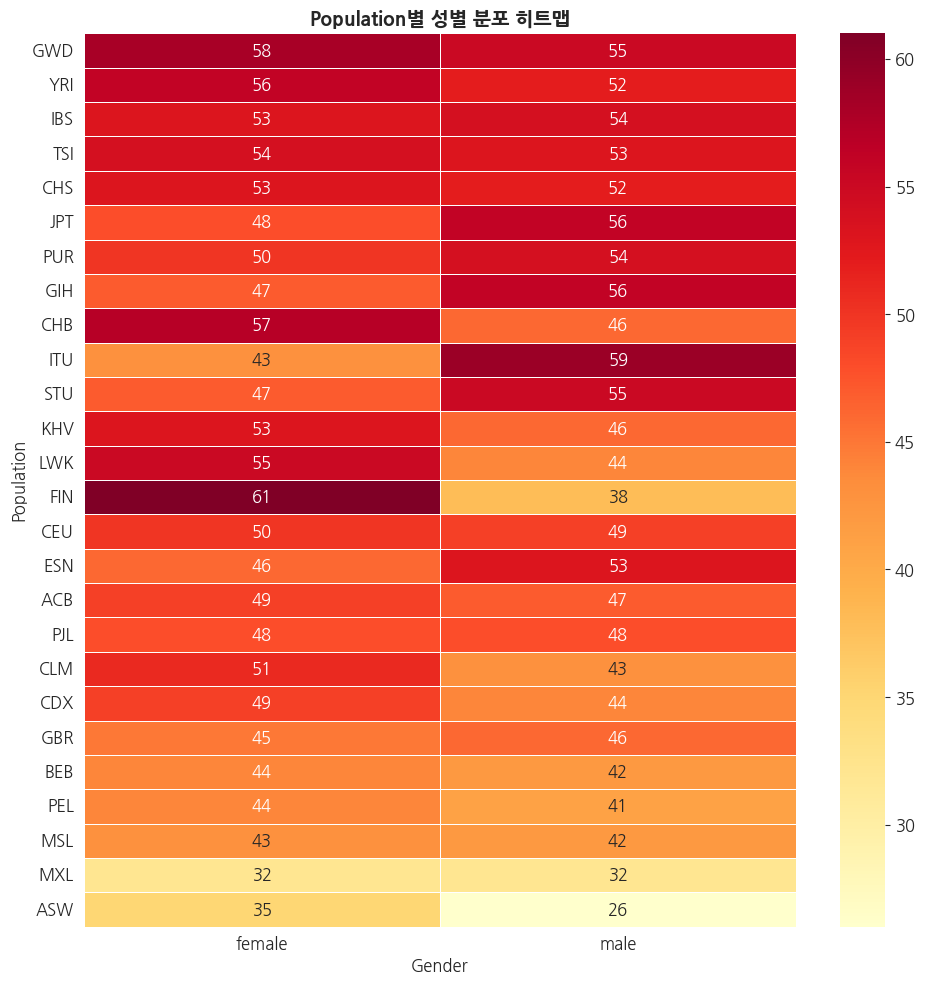
2.5 가계(Family) 구조 분석
python
# 가계 관계 분석
family_col = [c for c in ped.columns if 'amily' in c][0]
paternal_col = [c for c in ped.columns if 'aternal' in c][0]
maternal_col = [c for c in ped.columns if 'aternal' in c and 'M' in c[0]][0] if len([c for c in ped.columns if 'aternal' in c]) > 1 else [c for c in ped.columns if 'other' in c.lower() or 'maternal' in c.lower()][0]
# Relationship 분포
rel_col = [c for c in ped.columns if 'elation' in c.lower()][0]
fig, axes = plt.subplots(1, 2, figsize=(16, 6))
# Relationship 유형 분포
rel_counts = ped[rel_col].value_counts()
rel_counts.plot(kind='barh', ax=axes[0], color=sns.color_palette('Set2', len(rel_counts)), edgecolor='black')
axes[0].set_title('Relationship 유형별 분포', fontsize=14, fontweight='bold')
axes[0].set_xlabel('샘플 수')
# Trio 가족 식별 (부모 정보가 있는 샘플)
has_parents = ped[(ped[paternal_col] != '0') & (ped[paternal_col].notna())]
trio_count = len(has_parents)
unrelated_count = len(ped) - trio_count
axes[1].pie(
[unrelated_count, trio_count],
labels=['Unrelated', 'With Parents Info'],
autopct='%1.1f%%',
colors=['#3498db', '#e74c3c'],
startangle=90,
textprops={'fontsize': 12}
)
axes[1].set_title('부모 정보 유무', fontsize=14, fontweight='bold')
plt.tight_layout()
plt.show()
print(f'전체 샘플: {len(ped)}')
print(f'부모 정보 있는 샘플: {trio_count}')
print(f'Unrelated 샘플: {unrelated_count}')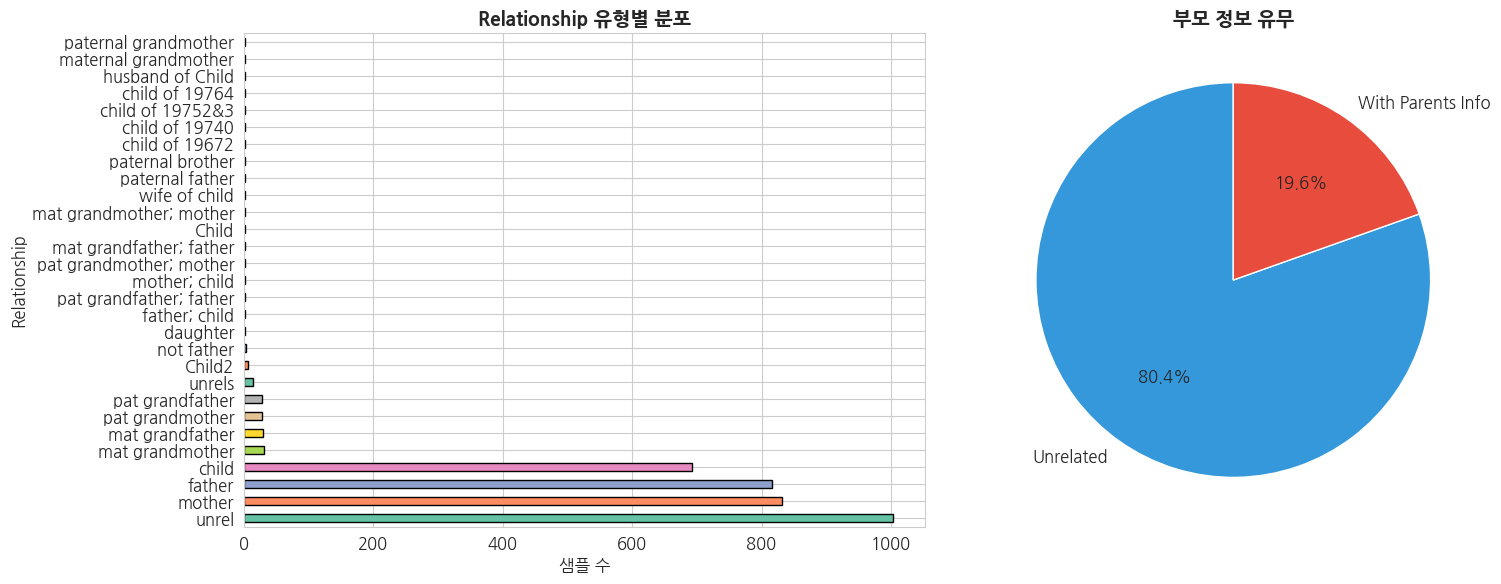
전체 샘플: 3501
부모 정보 있는 샘플: 685
Unrelated 샘플: 2816
2.6 Population 구조 요약 통계
python
# 포괄적 요약 통계 테이블 생성
summary_stats = panel.groupby('super_pop').agg(
total_samples=('sample', 'count'),
n_populations=('pop', 'nunique'),
n_male=('gender', lambda x: (x == 'male').sum()),
n_female=('gender', lambda x: (x == 'female').sum())
).reset_index()
summary_stats['male_ratio'] = (summary_stats['n_male'] / summary_stats['total_samples'] * 100).round(1)
summary_stats['female_ratio'] = (summary_stats['n_female'] / summary_stats['total_samples'] * 100).round(1)
# Super Population 설명 추가
sp_description = {
'AFR': 'African',
'AMR': 'Ad Mixed American',
'EAS': 'East Asian',
'EUR': 'European',
'SAS': 'South Asian'
}
summary_stats['description'] = summary_stats['super_pop'].map(sp_description)
print('=== Super Population별 요약 통계 ===')
display_cols = ['super_pop', 'description', 'total_samples', 'n_populations', 'n_male', 'n_female', 'male_ratio', 'female_ratio']
summary_stats[display_cols]text
=== Super Population별 요약 통계 ===| super_pop | description | total_samples | n_populations | n_male | n_female | male_ratio | female_ratio | |
|---|---|---|---|---|---|---|---|---|
| 0 | AFR | African | 661 | 7 | 319 | 342 | 48.3 | 51.7 |
| 1 | AMR | Ad Mixed American | 347 | 4 | 170 | 177 | 49.0 | 51.0 |
| 2 | EAS | East Asian | 504 | 5 | 244 | 260 | 48.4 | 51.6 |
| 3 | EUR | European | 503 | 5 | 240 | 263 | 47.7 | 52.3 |
| 4 | SAS | South Asian | 489 | 5 | 260 | 229 | 53.2 | 46.8 |
3. 유전변이(VCF)-임상 표현형 통합 분석을 위한 전처리
VCF 파일과 임상 표현형 데이터를 결합하기 위한 전처리를 수행합니다.
3.1 VCF 파일 샘플 목록 추출
python
import subprocess
# VCF 파일에서 샘플 목록 추출 (bcftools 사용)
vcf_path = f'{INDIVIDUAL_DIR}/ALL.chr22.phase3_shapeit2_mvncall_integrated_v5b.20130502.genotypes.vcf.gz'
result = subprocess.run(
['bcftools', 'query', '-l', vcf_path],
capture_output=True, text=True
)
vcf_samples = result.stdout.strip().split('\n')
print(f'VCF 파일 내 샘플 수: {len(vcf_samples)}')
print(f'처음 10개 샘플: {vcf_samples[:10]}')text
VCF 파일 내 샘플 수: 2504
처음 10개 샘플: ['HG00096', 'HG00097', 'HG00099', 'HG00100', 'HG00101', 'HG00102', 'HG00103', 'HG00105', 'HG00106', 'HG00107']python
# Panel 데이터와 VCF 샘플 간 매칭 확인
panel_samples = set(panel['sample'].values)
vcf_sample_set = set(vcf_samples)
common_samples = panel_samples & vcf_sample_set
only_panel = panel_samples - vcf_sample_set
only_vcf = vcf_sample_set - panel_samples
print(f'Panel에만 있는 샘플: {len(only_panel)}')
print(f'VCF에만 있는 샘플: {len(only_vcf)}')
print(f'공통 샘플: {len(common_samples)}')
print(f'\n→ Panel과 VCF의 샘플이 완전히 일치합니다.' if len(only_panel) == 0 and len(only_vcf) == 0 else f'\n→ 불일치 샘플이 있습니다.')text
Panel에만 있는 샘플: 0
VCF에만 있는 샘플: 0
공통 샘플: 2504
→ Panel과 VCF의 샘플이 완전히 일치합니다.3.2 분석용 통합 데이터셋 구축
VCF의 샘플 순서와 임상 정보를 정확히 매칭한 통합 데이터셋을 생성합니다.
python
# VCF 샘플 순서에 맞춰 panel 정보 정렬
vcf_sample_df = pd.DataFrame({'sample': vcf_samples, 'vcf_order': range(len(vcf_samples))})
# 통합 데이터셋 생성
integrated = vcf_sample_df.merge(panel, on='sample', how='left')
print(f'통합 데이터셋 크기: {integrated.shape}')
print(f'결측치 확인:')
print(integrated.isnull().sum())
print()
integrated.head(10)text
통합 데이터셋 크기: (2504, 7)
결측치 확인:
sample 0
vcf_order 0
pop 0
super_pop 0
gender 0
Unnamed: 4 2504
Unnamed: 5 2504
dtype: int64| sample | vcf_order | pop | super_pop | gender | Unnamed: 4 | Unnamed: 5 | |
|---|---|---|---|---|---|---|---|
| 0 | HG00096 | 0 | GBR | EUR | male | NaN | NaN |
| 1 | HG00097 | 1 | GBR | EUR | female | NaN | NaN |
| 2 | HG00099 | 2 | GBR | EUR | female | NaN | NaN |
| 3 | HG00100 | 3 | GBR | EUR | female | NaN | NaN |
| 4 | HG00101 | 4 | GBR | EUR | male | NaN | NaN |
| 5 | HG00102 | 5 | GBR | EUR | female | NaN | NaN |
| 6 | HG00103 | 6 | GBR | EUR | male | NaN | NaN |
| 7 | HG00105 | 7 | GBR | EUR | male | NaN | NaN |
| 8 | HG00106 | 8 | GBR | EUR | female | NaN | NaN |
| 9 | HG00107 | 9 | GBR | EUR | male | NaN | NaN |
3.3 분석 대상 서브셋 생성
특정 분석 목적에 맞는 서브셋을 미리 정의합니다.
python
# EAS (동아시아) 서브셋 - 한국인 관련 분석에 활용
eas_samples = integrated[integrated['super_pop'] == 'EAS']
print(f'=== EAS (동아시아) 서브셋 ===')
print(f'샘플 수: {len(eas_samples)}')
print(f'포함 Population: {eas_samples["pop"].unique()}')
print(f' - CDX: Chinese Dai in Xishuangbanna')
print(f' - CHB: Han Chinese in Beijing')
print(f' - CHS: Southern Han Chinese')
print(f' - JPT: Japanese in Tokyo')
print(f' - KHV: Kinh in Ho Chi Minh City')
print()
# Unrelated 샘플만 추출 (GWAS 분석용)
# Pedigree에서 관계 정보 확인하여 unrelated 샘플 필터링
ped_id_col = [c for c in ped.columns if 'ndividual' in c][0]
paternal_col = [c for c in ped.columns if 'aternal' in c][0]
unrelated_ids = ped[ped[paternal_col] == '0'][ped_id_col].values
unrelated_samples = integrated[integrated['sample'].isin(unrelated_ids)]
print(f'=== Unrelated 샘플 서브셋 (GWAS용) ===')
print(f'전체: {len(integrated)} → Unrelated: {len(unrelated_samples)}')text
=== EAS (동아시아) 서브셋 ===
샘플 수: 504
포함 Population: ['CHS' 'CDX' 'KHV' 'CHB' 'JPT']
- CDX: Chinese Dai in Xishuangbanna
- CHB: Han Chinese in Beijing
- CHS: Southern Han Chinese
- JPT: Japanese in Tokyo
- KHV: Kinh in Ho Chi Minh City
=== Unrelated 샘플 서브셋 (GWAS용) ===
전체: 2504 → Unrelated: 24953.4 개인 샘플 VCF에서 기본 변이 통계 미리보기
1명의 샘플을 선택하여 VCF에서 기본적인 변이 정보를 추출합니다. (2강에서 상세 분석 예정)
python
# 샘플 1명 선택 (EAS - JPT 일본인 샘플)
target_sample = eas_samples[eas_samples['pop'] == 'JPT']['sample'].values[0]
print(f'선택 샘플: {target_sample}')
print(f'Population: JPT (Japanese in Tokyo)')
print(f'Super Population: EAS (East Asian)')
print()
# bcftools로 해당 샘플의 변이 수 확인 (chr22 기준)
result = subprocess.run(
['bcftools', 'view', '-s', target_sample, '--min-ac', '1', '-H', vcf_path],
capture_output=True, text=True
)
variant_lines = result.stdout.strip().split('\n')
n_variants = len([l for l in variant_lines if l.strip()])
print(f'{target_sample}의 chr22 변이 수: {n_variants:,}')text
선택 샘플: NA18939
Population: JPT (Japanese in Tokyo)
Super Population: EAS (East Asian)
NA18939의 chr22 변이 수: 56,754python
# 해당 샘플의 변이 타입 미리보기 (처음 1000개만)
result = subprocess.run(
['bcftools', 'view', '-s', target_sample, '--min-ac', '1', vcf_path],
capture_output=True, text=True
)
# 변이 타입 분류
snp_count = 0
indel_count = 0
other_count = 0
for line in variant_lines[:5000]:
if not line.strip() or line.startswith('#'):
continue
fields = line.split('\t')
if len(fields) < 5:
continue
ref = fields[3]
alts = fields[4].split(',')
is_snp = all(len(alt) == 1 and len(ref) == 1 for alt in alts)
if is_snp:
snp_count += 1
else:
indel_count += 1
print(f'=== {target_sample} chr22 변이 타입 미리보기 (처음 5000개) ===')
print(f'SNP: {snp_count:,}')
print(f'INDEL: {indel_count:,}')
print(f'\n→ 2강에서 VCF 구조 및 변이 타입에 대해 상세히 분석합니다.')text
=== NA18939 chr22 변이 타입 미리보기 (처음 5000개) ===
SNP: 4,354
INDEL: 646
→ 2강에서 VCF 구조 및 변이 타입에 대해 상세히 분석합니다.3.5 전처리된 데이터 저장
후속 강의에서 사용할 수 있도록 통합 데이터를 저장합니다.
python
# 통합 임상 데이터 저장
output_path = f'{DATA_DIR}/phenotype/integrated_sample_info.tsv'
integrated.to_csv(output_path, sep='\t', index=False)
print(f'통합 데이터 저장 완료: {output_path}')
print(f' - 샘플 수: {len(integrated)}')
print(f' - 컬럼: {list(integrated.columns)}')
# EAS 서브셋 저장
eas_output = f'{DATA_DIR}/phenotype/eas_sample_info.tsv'
eas_samples.to_csv(eas_output, sep='\t', index=False)
print(f'\nEAS 서브셋 저장 완료: {eas_output}')
print(f' - 샘플 수: {len(eas_samples)}')
# Unrelated 샘플 목록 저장 (GWAS용)
unrelated_output = f'{DATA_DIR}/phenotype/unrelated_samples.txt'
unrelated_samples[['sample']].to_csv(unrelated_output, sep='\t', index=False, header=False)
print(f'\nUnrelated 샘플 목록 저장 완료: {unrelated_output}')
print(f' - 샘플 수: {len(unrelated_samples)}')text
통합 데이터 저장 완료: /tier4/DSC/jheepark/bbp-wgs-data/phenotype/integrated_sample_info.tsv
- 샘플 수: 2504
- 컬럼: ['sample', 'vcf_order', 'pop', 'super_pop', 'gender', 'Unnamed: 4', 'Unnamed: 5']
EAS 서브셋 저장 완료: /tier4/DSC/jheepark/bbp-wgs-data/phenotype/eas_sample_info.tsv
- 샘플 수: 504
Unrelated 샘플 목록 저장 완료: /tier4/DSC/jheepark/bbp-wgs-data/phenotype/unrelated_samples.txt
- 샘플 수: 24954. 요약 및 다음 강의 안내
이번 강의 요약
| 항목 | 내용 |
|---|---|
| 데이터 소스 | 1000 Genomes Project Phase 3 |
| 전체 샘플 | 2,504명 |
| Super Population | 5개 (AFR, AMR, EAS, EUR, SAS) |
| 세부 Population | 26개 |
| 주요 분석 | 인구집단 분포, 성별 연관성(χ²), 가계 구조 |
| 전처리 산출물 | 통합 데이터셋, EAS 서브셋, Unrelated 샘플 목록 |
핵심 포인트
- WGS 분석 전 임상/표현형 데이터의 구조를 이해하는 것이 중요합니다.
- 인구집단 구성, 성별 분포, 가계 관계 등은 후속 유전 분석의 교란 요인이 될 수 있습니다.
- VCF 데이터와 임상 정보의 샘플 ID 매칭을 반드시 확인해야 합니다.
- 분석 목적에 따라 적절한 서브셋 (예: Unrelated 샘플) 을 선별하는 것이 필요합니다.
다음 강의 예고
2강: VCF 기초 통계 분석
- VCF 파일의 주요 컬럼 확인 및 통계 지표 계산
- SNP·INDEL 등 변이 타입 분포 시각화
- Mutation Profile 기반 변이 특성 요약
- 필터링 조건별 변이 수 비교 분석