4강: 환자 코호트 기반 질병 연관성 분석
BBP WGS 분석 튜토리얼
학습 목표
- 다수 환자 VCF 통합 및 코호트 QC 수행 - Joint VCF 품질 검증 및 변이/샘플 필터링
- GWAS 분석 실행 및 결과 시각화 - 표현형-변이 연관성 분석 (Manhattan plot, QQ plot)
- PRS(Polygenic Risk Score) 계산 - 개인별 유전적 위험도 점수 산출 및 비교
사용 데이터
데이터 루트: /tier4/DSC/jheepark/bbp-wgs-data/
| 데이터 | 경로 | 설명 |
|---|---|---|
| 1000 Genomes chr22 Joint VCF | individual/ALL.chr22.phase3_shapeit2_mvncall_integrated_v5b.20130502.genotypes.vcf.gz | 2,504 샘플 joint VCF |
| 통합 샘플 정보 | phenotype/integrated_sample_info.tsv | 1강에서 생성한 Panel + Pedigree 병합 데이터 |
| Unrelated 샘플 목록 | phenotype/unrelated_samples.txt | 부모 정보 없는 unrelated 샘플 ID (GWAS용) |
Phenotype 은 EAS super population 여부 (case=EAS, control=그 외) 로 시뮬레이션하여 GWAS/PRS 를 수행합니다. 실제 운영에서는 PGS Catalog 의 weight 파일을 사용합니다.
산출물 (본 강의에서 생성)
| 파일 | 경로 | 설명 |
|---|---|---|
| 코호트 샘플 목록 | cohort/cohort_samples.txt | QC 통과 샘플 ID 리스트 |
| 코호트 VCF | cohort/cohort_chr22.vcf.gz | Unrelated × biallelic SNP × MAF≥0.01 × missing<5% 필터링된 VCF |
| PRS Weight 파일 | cohort/prs_weights.txt | GWAS 결과 기반 SNP weight 테이블 |
| PRS 결과 | cohort/cohort_prs.tsv | 샘플별 PRS raw / z-score |
사용 도구
- bcftools, vcftools (VCF QC)
- PLINK (명령어 예시, 통계 분석 일부는 Python으로 대체)
- PRSice-2 (명령어 예시)
- Python: cyvcf2, pandas, numpy, scipy, matplotlib, seaborn
주의
본 실습 환경은 PLINK/PRSice-2 가 설치되어 있지 않을 수 있습니다. 핵심 알고리즘을 Python 으로 구현하여 학습하고, 실제 운영 환경에서 사용할 PLINK/PRSice-2 명령어를 참고용 코드로 함께 제공합니다.
0. 환경 설정
python
import os
import subprocess
from collections import Counter, defaultdict
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
from scipy import stats
from cyvcf2 import VCF
from matplotlib import font_manager, rc
import warnings
warnings.filterwarnings('ignore')
# 시각화 설정 (seaborn 스타일 먼저 → 한글 폰트 뒤에 적용)
sns.set_style('whitegrid')
sns.set_palette('Set2')
font_path = '/usr/share/fonts/truetype/nanum/NanumGothic.ttf'
font_manager.fontManager.addfont(font_path)
rc('font', family='NanumGothic')
plt.rcParams['axes.unicode_minus'] = False
plt.rcParams['figure.figsize'] = (12, 6)
plt.rcParams['font.size'] = 12
DATA_DIR = '/tier4/DSC/jheepark/bbp-wgs-data'
INDIVIDUAL_DIR = f'{DATA_DIR}/individual'
PHENOTYPE_DIR = f'{DATA_DIR}/phenotype'
COHORT_DIR = f'{DATA_DIR}/cohort'
os.makedirs(COHORT_DIR, exist_ok=True)
VCF_PATH = f'{INDIVIDUAL_DIR}/ALL.chr22.phase3_shapeit2_mvncall_integrated_v5b.20130502.genotypes.vcf.gz'
print('환경 설정 완료!')
print(f'Cohort VCF: {VCF_PATH}')text
환경 설정 완료!
Cohort VCF: /tier4/DSC/jheepark/bbp-wgs-data/individual/ALL.chr22.phase3_shapeit2_mvncall_integrated_v5b.20130502.genotypes.vcf.gz1. 코호트 VCF 통합 및 QC
1.1 Joint VCF 의 개념
Joint calling 은 여러 샘플을 동시에 variant calling 하여 생성하는 멀티-샘플 VCF입니다. 이번 실습의 1000 Genomes chr22 VCF는 이미 2,504명의 joint VCF 입니다.
| 항목 | 개인 VCF | Joint VCF |
|---|---|---|
| 샘플 수 | 1명 | 수백~수만명 |
| 변이 수 | 샘플 보유 변이만 | 합집합 (누군가 1명이라도 보유) |
| Missing genotype | 없음 | 샘플별 발생 가능 |
| 주요 QC 대상 | 개인 변이 품질 | 변이 × 샘플 quality |
python
# Joint VCF 기본 정보 확인
result = subprocess.run(
['bcftools', 'stats', VCF_PATH],
capture_output=True, text=True
)
stats_output = result.stdout
# 주요 통계 추출
print('=== Joint VCF 기본 통계 ===')
for line in stats_output.split('\n'):
if line.startswith('SN'):
print(line)
# 샘플 수
r = subprocess.run(['bcftools', 'query', '-l', VCF_PATH], capture_output=True, text=True)
samples = r.stdout.strip().split('\n')
print(f'\n총 샘플 수: {len(samples):,}')text
=== Joint VCF 기본 통계 ===
SN 0 number of samples: 2504
SN 0 number of records: 1103547
SN 0 number of no-ALTs: 0
SN 0 number of SNPs: 1060388
SN 0 number of MNPs: 0
SN 0 number of indels: 43230
SN 0 number of others: 801
SN 0 number of multiallelic sites: 6348
SN 0 number of multiallelic SNP sites: 4063
총 샘플 수: 2,5041.2 Unrelated 샘플만 남기기 (코호트 QC 1단계)
GWAS 는 통계적으로 독립적인 샘플이 필요하므로, 1강에서 생성한 unrelated 샘플 목록을 기반으로 코호트를 구성합니다.
python
# Unrelated 샘플 목록 로드
unrelated_df = pd.read_csv(f'{PHENOTYPE_DIR}/unrelated_samples.txt', sep='\t', header=None, names=['sample'])
integrated = pd.read_csv(f'{PHENOTYPE_DIR}/integrated_sample_info.tsv', sep='\t')
print(f'Unrelated 샘플 수: {len(unrelated_df):,}')
print(f'통합 데이터 샘플 수: {len(integrated):,}')
# VCF 샘플 중 unrelated 샘플만 추출
common = set(samples) & set(unrelated_df['sample']) & set(integrated['sample'])
print(f'\n코호트 구성 샘플 (VCF ∩ Unrelated ∩ Phenotype): {len(common):,}')
# Super Population 별 분포
cohort_info = integrated[integrated['sample'].isin(common)]
print('\n=== 코호트 Super Population 분포 ===')
print(cohort_info['super_pop'].value_counts())text
Unrelated 샘플 수: 2,495
통합 데이터 샘플 수: 2,504
코호트 구성 샘플 (VCF ∩ Unrelated ∩ Phenotype): 2,495
=== 코호트 Super Population 분포 ===
super_pop
AFR 658
EAS 503
EUR 499
SAS 488
AMR 347
Name: count, dtype: int641.3 코호트 VCF 추출 (bcftools view)
PLINK 입력으로 쓰기 편하게 다음 기준을 동시에 적용한 VCF 를 만듭니다.
- Unrelated 샘플만
- Biallelic SNP 만
- MAF ≥ 0.01
- Missingness < 5%
python
# Unrelated 샘플 리스트 파일 저장
cohort_samples_file = f'{COHORT_DIR}/cohort_samples.txt'
cohort_info['sample'].to_csv(cohort_samples_file, index=False, header=False)
print(f'코호트 샘플 리스트 저장: {cohort_samples_file} ({len(cohort_info):,}명)')
# 코호트 VCF 생성 (필터링 포함)
cohort_vcf = f'{COHORT_DIR}/cohort_chr22.vcf.gz'
cmd = [
'bcftools', 'view',
'-S', cohort_samples_file,
'-v', 'snps',
'-m2', '-M2', # biallelic only
'-q', '0.01:minor', # MAF >= 0.01
'-e', 'F_MISSING > 0.05',# missingness 제외
'--force-samples',
'-Oz', '-o', cohort_vcf,
VCF_PATH
]
result = subprocess.run(cmd, capture_output=True, text=True)
print(f'VCF 생성 rc={result.returncode}')
subprocess.run(['tabix', '-p', 'vcf', '-f', cohort_vcf],
capture_output=True, text=True)
# 결과 확인
r = subprocess.run(['bcftools', 'view', '-H', cohort_vcf],
capture_output=True, text=True)
n_cohort_variants = len([l for l in r.stdout.strip().split('\n') if l])
print(f'\n=== 코호트 QC 결과 ===')
print(f'샘플 수 : {len(cohort_info):,}')
print(f'변이 수 : {n_cohort_variants:,} (필터링 후)')
print(f'출력 파일 : {cohort_vcf}')text
코호트 샘플 리스트 저장: /tier4/DSC/jheepark/bbp-wgs-data/cohort/cohort_samples.txt (2,495명)
VCF 생성 rc=0
=== 코호트 QC 결과 ===
샘플 수 : 2,495
변이 수 : 171,895 (필터링 후)
출력 파일 : /tier4/DSC/jheepark/bbp-wgs-data/cohort/cohort_chr22.vcf.gz1.4 QC 지표 계산: MAF, Missingness, HWE
Hardy-Weinberg Equilibrium (HWE) 는 "큰 집단에서 임의 교배 시 allele frequency가 세대간 유지된다"는 법칙입니다. HWE에서 크게 벗어나는 변이는 genotyping 오류 가능성이 높아 GWAS 전 제거합니다.
$$\chi^2 = \sum \frac{(Observed - Expected)^2}{Expected}$$
여기서 Expected 는 allele frequency $p$, $q$ 로부터 $p^2, 2pq, q^2$ 로 계산합니다.
python
# cyvcf2 로 일부 변이(처음 50,000개)에 대해 QC 지표 계산
vcf = VCF(cohort_vcf)
qc_records = []
max_variants = 50000 # 실습 속도를 위해 제한
for i, v in enumerate(vcf):
if i >= max_variants:
break
gts = v.genotypes # list of [a1, a2, phased]
n_total = len(gts)
n_missing = 0
n_hom_ref = 0
n_het = 0
n_hom_alt = 0
for gt in gts:
a1, a2 = gt[0], gt[1]
if a1 < 0 or a2 < 0:
n_missing += 1
else:
alt = (a1 == 1) + (a2 == 1)
if alt == 0:
n_hom_ref += 1
elif alt == 1:
n_het += 1
else:
n_hom_alt += 1
n_called = n_hom_ref + n_het + n_hom_alt
if n_called == 0:
continue
# Allele Frequency
af = (n_het + 2 * n_hom_alt) / (2 * n_called)
maf = min(af, 1 - af)
missingness = n_missing / n_total
# HWE Chi-square (biallelic 가정)
p = 1 - af # ref allele freq
q = af
e_hom_ref = n_called * p * p
e_het = n_called * 2 * p * q
e_hom_alt = n_called * q * q
if min(e_hom_ref, e_het, e_hom_alt) > 0:
chi2 = ((n_hom_ref - e_hom_ref) ** 2 / e_hom_ref
+ (n_het - e_het) ** 2 / e_het
+ (n_hom_alt- e_hom_alt)** 2 / e_hom_alt)
hwe_p = 1 - stats.chi2.cdf(chi2, df=1)
else:
chi2, hwe_p = np.nan, np.nan
qc_records.append({
'CHROM': v.CHROM, 'POS': v.POS, 'ID': v.ID,
'REF': v.REF, 'ALT': v.ALT[0] if v.ALT else '.',
'AF': af, 'MAF': maf,
'MISSING': missingness,
'N_HET': n_het, 'N_HOM_REF': n_hom_ref, 'N_HOM_ALT': n_hom_alt,
'HWE_CHI2': chi2, 'HWE_P': hwe_p,
})
vcf.close()
qc_df = pd.DataFrame(qc_records)
print(f'QC 지표 계산 완료: {len(qc_df):,} 변이')
qc_df.head()text
QC 지표 계산 완료: 50,000 변이| CHROM | POS | ID | REF | ALT | AF | MAF | MISSING | N_HET | N_HOM_REF | N_HOM_ALT | HWE_CHI2 | HWE_P | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 22 | 16051249 | None | T | C | 0.112625 | 0.112625 | 0.0 | 454 | 1987 | 54 | 20.048800 | 7.549087e-06 |
| 1 | 22 | 16052080 | None | G | A | 0.141283 | 0.141283 | 0.0 | 705 | 1790 | 0 | 67.537834 | 2.220446e-16 |
| 2 | 22 | 16052463 | None | T | C | 0.010220 | 0.010220 | 0.0 | 47 | 2446 | 2 | 11.849542 | 5.767554e-04 |
| 3 | 22 | 16052684 | None | A | C | 0.034068 | 0.034068 | 0.0 | 158 | 2331 | 6 | 3.566494 | 5.895668e-02 |
| 4 | 22 | 16052962 | None | C | T | 0.093988 | 0.093988 | 0.0 | 393 | 2064 | 38 | 14.079035 | 1.752867e-04 |
python
# QC 지표 시각화
fig, axes = plt.subplots(2, 2, figsize=(16, 10))
# 1. MAF 분포
axes[0, 0].hist(qc_df['MAF'], bins=50, color='#3498db', edgecolor='black', linewidth=0.3)
axes[0, 0].axvline(0.05, color='red', linestyle='--', label='MAF=0.05')
axes[0, 0].set_title('MAF 분포', fontsize=13, fontweight='bold')
axes[0, 0].set_xlabel('Minor Allele Frequency')
axes[0, 0].set_ylabel('변이 수')
axes[0, 0].legend()
# 2. Missingness 분포
axes[0, 1].hist(qc_df['MISSING'], bins=50, color='#e74c3c', edgecolor='black', linewidth=0.3)
axes[0, 1].set_title('Missingness 분포', fontsize=13, fontweight='bold')
axes[0, 1].set_xlabel('Missing rate')
axes[0, 1].set_ylabel('변이 수')
axes[0, 1].set_yscale('log')
# 3. HWE p-value 분포 (-log10)
hwe_p = qc_df['HWE_P'].dropna()
hwe_p = hwe_p[hwe_p > 0]
neg_log_p = -np.log10(hwe_p)
axes[1, 0].hist(neg_log_p, bins=50, color='#9b59b6', edgecolor='black', linewidth=0.3)
axes[1, 0].axvline(-np.log10(1e-6), color='red', linestyle='--', label='p=1e-6')
axes[1, 0].set_title('HWE -log10(p) 분포', fontsize=13, fontweight='bold')
axes[1, 0].set_xlabel('-log10(HWE p-value)')
axes[1, 0].set_ylabel('변이 수')
axes[1, 0].legend()
# 4. MAF vs Missingness scatter
sample_idx = np.random.choice(len(qc_df), min(5000, len(qc_df)), replace=False)
axes[1, 1].scatter(qc_df['MAF'].iloc[sample_idx],
qc_df['MISSING'].iloc[sample_idx],
alpha=0.3, s=8, c='#2ecc71')
axes[1, 1].set_title('MAF vs Missingness', fontsize=13, fontweight='bold')
axes[1, 1].set_xlabel('MAF')
axes[1, 1].set_ylabel('Missingness')
plt.tight_layout()
plt.show()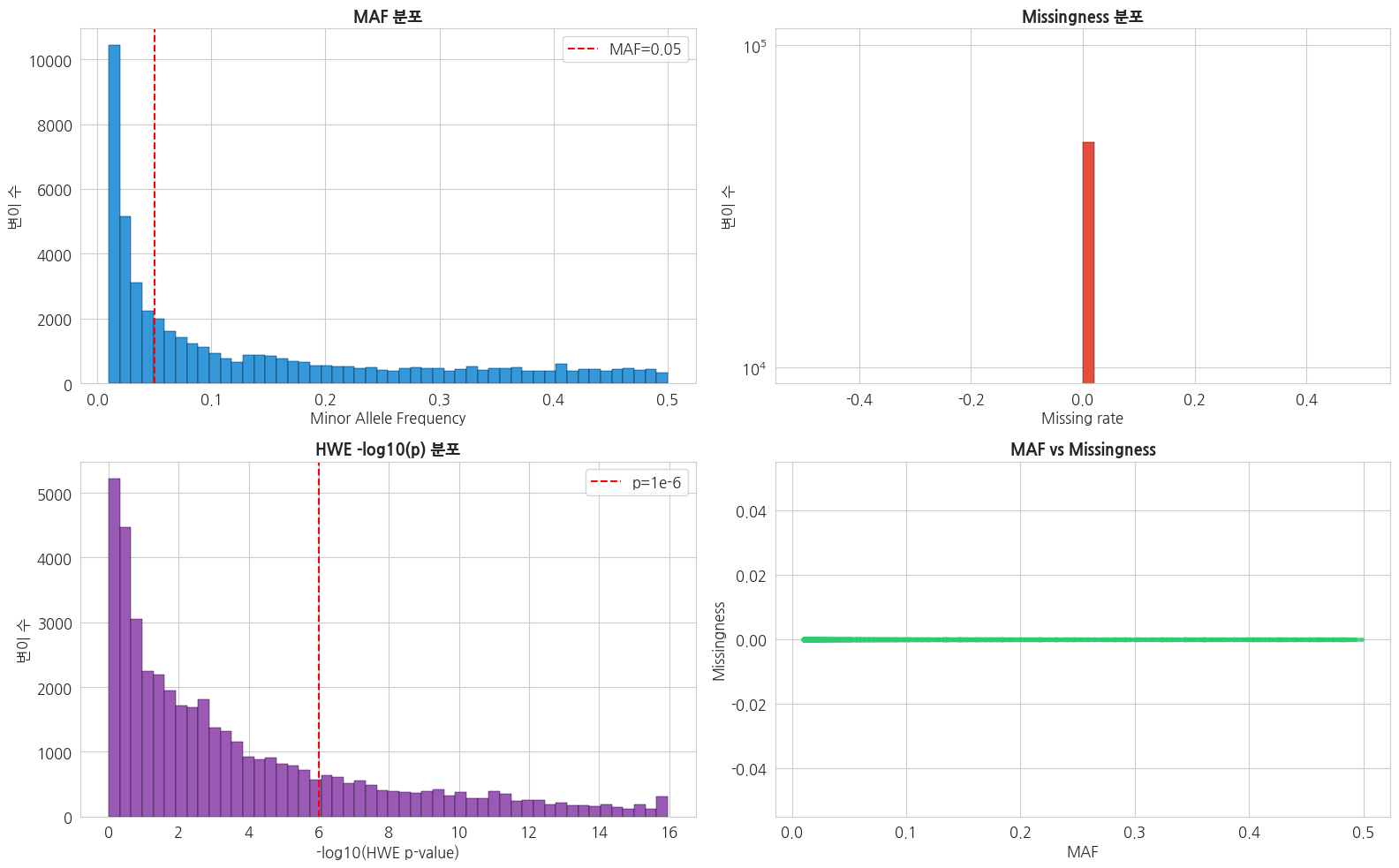
1.5 QC 통과 변이 요약
python
# QC 기준별 통과 변이 수
qc_steps = {
'0. 전체 (MAF≥0.01 필터 후)': len(qc_df),
'1. Missing < 5%': (qc_df['MISSING'] < 0.05).sum(),
'2. HWE p > 1e-6': (qc_df['HWE_P'] > 1e-6).sum(),
'3. MAF ≥ 0.05': (qc_df['MAF'] >= 0.05).sum(),
'4. 모든 QC 통과': ((qc_df['MISSING'] < 0.05) &
(qc_df['HWE_P'] > 1e-6) &
(qc_df['MAF'] >= 0.05)).sum(),
}
print('=== QC 단계별 통과 변이 수 ===')
for name, count in qc_steps.items():
print(f' {name:30s}: {count:>8,}')
# QC 통과 변이만 필터링
qc_pass = qc_df[(qc_df['MISSING'] < 0.05) &
(qc_df['HWE_P'] > 1e-6) &
(qc_df['MAF'] >= 0.01)].copy()
print(f'\n최종 GWAS 대상 변이: {len(qc_pass):,}')text
=== QC 단계별 통과 변이 수 ===
0. 전체 (MAF≥0.01 필터 후) : 50,000
1. Missing < 5% : 50,000
2. HWE p > 1e-6 : 33,697
3. MAF ≥ 0.05 : 28,872
4. 모든 QC 통과 : 16,803
최종 GWAS 대상 변이: 33,6971.6 PLINK 포맷 변환 명령어 (참고)
PLINK 는 GWAS 의 표준 도구입니다. VCF → BED/BIM/FAM 변환 명령어를 제공합니다.
bash
# VCF → PLINK binary (BED/BIM/FAM)
plink2 \
--vcf cohort_chr22.vcf.gz \
--make-bed \
--out cohort_chr22 \
--keep-allele-order
# 코호트 QC (PLINK)
plink2 \
--bfile cohort_chr22 \
--maf 0.01 \
--geno 0.05 \
--hwe 1e-6 \
--mind 0.05 \
--make-bed --out cohort_chr22_qc| PLINK 옵션 | 의미 |
|---|---|
--maf 0.01 | MAF < 0.01 변이 제거 |
--geno 0.05 | Missing rate > 5% 변이 제거 |
--hwe 1e-6 | HWE p < 1e-6 변이 제거 |
--mind 0.05 | 샘플 missing rate > 5% 샘플 제거 |
2. GWAS(Genome-Wide Association Study) 분석
2.1 GWAS 의 기본 원리
GWAS 는 각 변이마다 환자군-대조군의 allele 빈도 차이를 독립적으로 검정하여 연관성이 있는 변이를 찾는 분석입니다.
Case-control binary trait
- $2 \times 3$ 분할표 (genotype × case/control)
- Logistic regression: $\log\frac{p}{1-p} = \beta_0 + \beta_1 \cdot g + \mathbf{\beta^T X_{cov}}$
- 유의 기준: $p < 5 \times 10^{-8}$ (Bonferroni 보정)
이번 실습의 시뮬레이션
실제 1000 Genomes 샘플에는 임상 표현형이 없으므로, "EAS 집단 여부 = phenotype" 으로 가정하여 GWAS 를 시뮬레이션합니다. 이는 EAS 특이 allele frequency 변이를 찾는 인구집단 분화 변이 탐색과 같은 개념입니다.
python
# Phenotype 시뮬레이션: EAS=1 (case), 나머지=0 (control)
pheno = cohort_info[['sample', 'super_pop']].copy()
pheno['phenotype'] = (pheno['super_pop'] == 'EAS').astype(int)
pheno = pheno.set_index('sample')
print('=== Phenotype 분포 ===')
print(pheno['phenotype'].value_counts())
print(f'\n Case (EAS) : {(pheno["phenotype"]==1).sum():,}명')
print(f' Control (기타): {(pheno["phenotype"]==0).sum():,}명')text
=== Phenotype 분포 ===
phenotype
0 1992
1 503
Name: count, dtype: int64
Case (EAS) : 503명
Control (기타): 1,992명2.2 Python 으로 GWAS 구현 (Chi-square test)
각 변이에 대해 genotype×phenotype 2×3 분할표를 만들고 카이제곱 검정을 수행합니다.
python
# 샘플 순서 확인 및 phenotype 벡터 정렬
vcf = VCF(cohort_vcf)
vcf_samples = vcf.samples
pheno_vec = np.array([pheno.loc[s, 'phenotype'] if s in pheno.index else -1
for s in vcf_samples])
mask = pheno_vec >= 0
print(f'VCF 샘플: {len(vcf_samples)}, Phenotype 매칭: {mask.sum()}')
# GWAS 수행 (처음 50,000 변이)
gwas_records = []
n_tests = 0
for i, v in enumerate(vcf):
if i >= 50000:
break
# genotype count (alt dosage)
gts = v.genotypes
dosage = np.array([
(a1 == 1) + (a2 == 1) if a1 >= 0 and a2 >= 0 else -1
for a1, a2, _ in gts
])
valid = (dosage >= 0) & mask
if valid.sum() < 100:
continue
d = dosage[valid]
y = pheno_vec[valid]
# 2x3 contingency table
table = np.zeros((2, 3), dtype=int)
for g in [0, 1, 2]:
table[0, g] = ((y == 0) & (d == g)).sum()
table[1, g] = ((y == 1) & (d == g)).sum()
# Drop zero-column if monomorphic
col_sums = table.sum(axis=0)
if (col_sums > 0).sum() < 2:
continue
keep_cols = col_sums > 0
tbl = table[:, keep_cols]
try:
chi2, p, dof, _ = stats.chi2_contingency(tbl)
except ValueError:
continue
# Allele frequency per group (case/control)
case_af = (table[1, 1] + 2*table[1, 2]) / (2 * y.sum()) if y.sum() else 0
control_af = (table[0, 1] + 2*table[0, 2]) / (2 * (y==0).sum()) if (y==0).sum() else 0
gwas_records.append({
'CHROM': v.CHROM, 'POS': v.POS, 'ID': v.ID,
'REF': v.REF, 'ALT': v.ALT[0] if v.ALT else '.',
'CASE_AF': case_af, 'CTRL_AF': control_af,
'CHI2': chi2, 'P': p, 'DOF': dof,
})
n_tests += 1
vcf.close()
gwas_df = pd.DataFrame(gwas_records)
print(f'\nGWAS 완료: {len(gwas_df):,} 변이 테스트')
# 유의 수준
sig_threshold = 5e-8
n_suggest = 1e-5
print(f'\n유의 변이 (p < {sig_threshold:.0e}): {(gwas_df["P"] < sig_threshold).sum():,}')
print(f'Suggestive (p < {n_suggest:.0e}): {(gwas_df["P"] < n_suggest).sum():,}')text
VCF 샘플: 2495, Phenotype 매칭: 2495
GWAS 완료: 50,000 변이 테스트
유의 변이 (p < 5e-08): 25,593
Suggestive (p < 1e-05): 31,5342.3 Manhattan Plot
각 변이의 chromosome × position 위치에 $-\log_{10}(p)$ 를 그립니다. 유의 변이는 상단에 peak 로 나타납니다.
python
# Manhattan plot
gwas_df['neg_log_p'] = -np.log10(gwas_df['P'].clip(lower=1e-300))
fig, ax = plt.subplots(figsize=(16, 6))
# chr22 단일 염색체이므로 X축은 position (Mb)
pos_mb = gwas_df['POS'] / 1e6
ax.scatter(pos_mb, gwas_df['neg_log_p'],
s=8, c=gwas_df['neg_log_p'], cmap='viridis',
alpha=0.6)
ax.axhline(-np.log10(5e-8), color='red', linestyle='--',
linewidth=1.5, label='Genome-wide significance (5e-8)')
ax.axhline(-np.log10(1e-5), color='orange', linestyle='--',
linewidth=1.2, label='Suggestive (1e-5)')
ax.set_xlabel('Chromosome 22 Position (Mb)', fontsize=12)
ax.set_ylabel('-log10(p-value)', fontsize=12)
ax.set_title('Manhattan Plot - EAS vs Non-EAS GWAS (chr22)',
fontsize=14, fontweight='bold')
ax.legend(loc='upper right')
plt.tight_layout()
plt.show()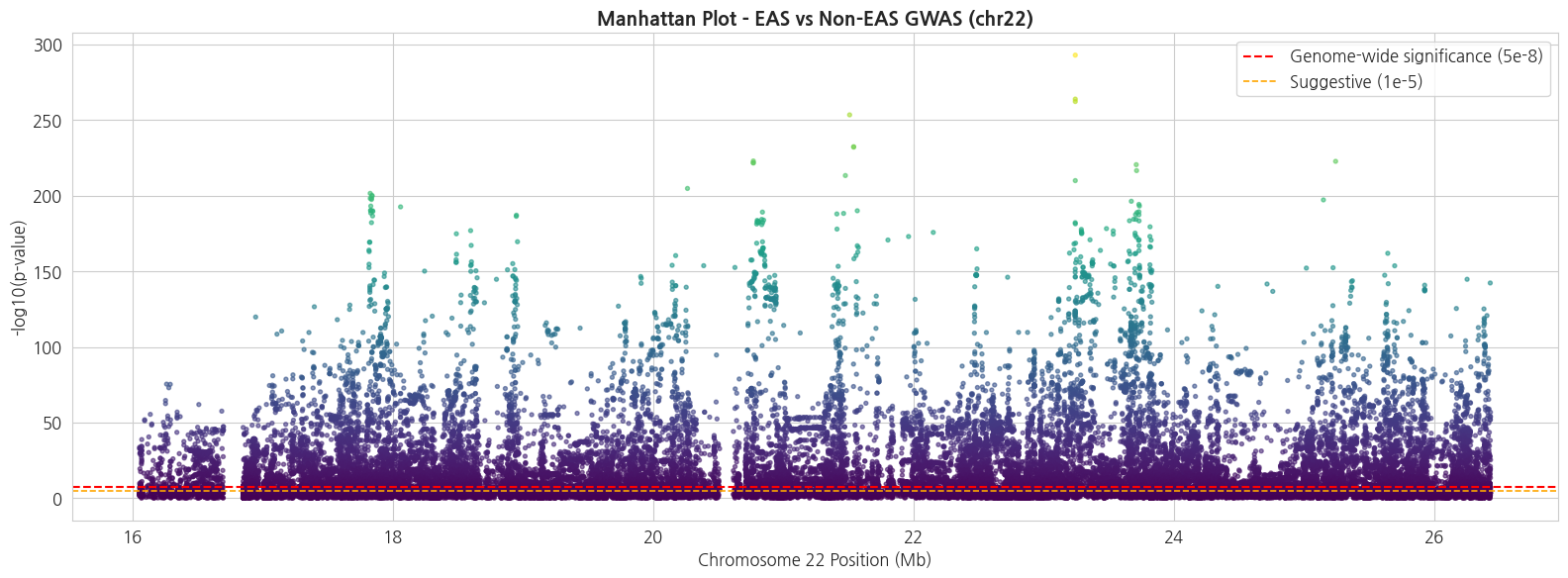
2.4 QQ Plot 및 Genomic Inflation Factor (λ)
QQ plot 은 관측된 p-value 와 기대 p-value 의 일치도를 보여줍니다. Genomic Inflation Factor (λ) 는 기대값 대비 분포 부풀림 정도를 평가합니다.
$$\lambda = \frac{\text{median}(\chi^2_{obs})}{\chi^2_{0.5, 1df}}$$
- $\lambda \approx 1.0$ : 이상적
- $\lambda > 1.1$ : 인구구조 등에 의한 inflation 의심
python
# QQ plot
fig, axes = plt.subplots(1, 2, figsize=(14, 6))
# 1. QQ plot
p_sorted = np.sort(gwas_df['P'].values)
expected = -np.log10((np.arange(1, len(p_sorted) + 1) - 0.5) / len(p_sorted))
observed = -np.log10(p_sorted)
axes[0].scatter(expected, observed, s=6, alpha=0.5, c='#3498db')
lim = max(expected.max(), observed.max())
axes[0].plot([0, lim], [0, lim], 'r--', linewidth=1)
axes[0].set_xlabel('Expected -log10(p)')
axes[0].set_ylabel('Observed -log10(p)')
axes[0].set_title('QQ Plot', fontsize=13, fontweight='bold')
# Genomic Inflation Factor
lambda_gc = np.median(gwas_df['CHI2']) / stats.chi2.ppf(0.5, df=1)
axes[0].text(0.05, 0.95, f'λ = {lambda_gc:.3f}',
transform=axes[0].transAxes, fontsize=14,
verticalalignment='top',
bbox=dict(boxstyle='round', facecolor='wheat', alpha=0.8))
# 2. Case vs Control AF scatter (상위 유의 변이 highlight)
axes[1].scatter(gwas_df['CTRL_AF'], gwas_df['CASE_AF'],
s=6, alpha=0.3, c='grey', label='전체 변이')
sig_variants = gwas_df[gwas_df['P'] < 5e-8]
axes[1].scatter(sig_variants['CTRL_AF'], sig_variants['CASE_AF'],
s=20, alpha=0.8, c='red',
label=f'유의 변이 (p<5e-8, n={len(sig_variants)})')
axes[1].plot([0, 1], [0, 1], 'k--', linewidth=0.8)
axes[1].set_xlabel('Control Allele Frequency')
axes[1].set_ylabel('Case Allele Frequency')
axes[1].set_title('Case vs Control Allele Frequency', fontsize=13, fontweight='bold')
axes[1].legend()
plt.tight_layout()
plt.show()
print(f'Genomic Inflation Factor λ = {lambda_gc:.4f}')
if lambda_gc > 1.1:
print(' ※ λ > 1.1: 인구구조(population stratification) 등에 의한 inflation 의심')
print(' ※ 실제 GWAS 에서는 Principal Component 를 covariate 로 넣어 보정합니다.')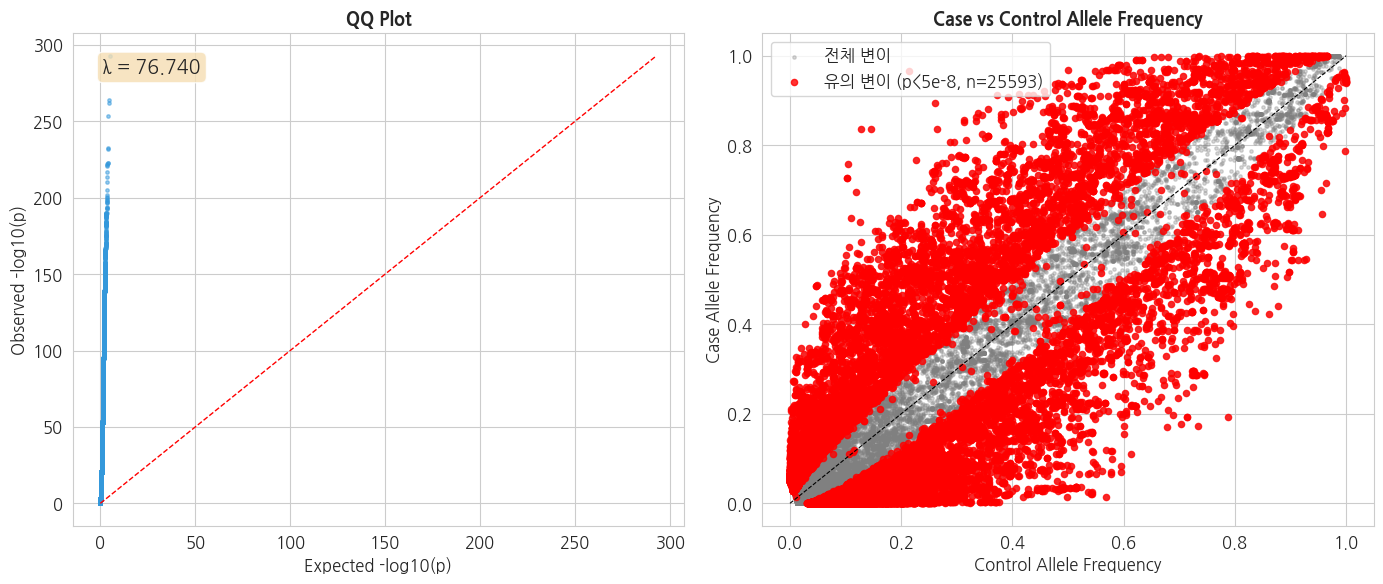
Genomic Inflation Factor λ = 76.7401
※ λ > 1.1: 인구구조(population stratification) 등에 의한 inflation 의심
※ 실제 GWAS 에서는 Principal Component 를 covariate 로 넣어 보정합니다.
2.5 상위 유의 변이 확인
python
# 상위 20 유의 변이
top_hits = gwas_df.nsmallest(20, 'P')[['CHROM', 'POS', 'ID', 'REF', 'ALT',
'CASE_AF', 'CTRL_AF', 'P', 'CHI2']]
top_hits['P'] = top_hits['P'].map(lambda p: f'{p:.2e}')
top_hits['CASE_AF'] = top_hits['CASE_AF'].round(3)
top_hits['CTRL_AF'] = top_hits['CTRL_AF'].round(3)
top_hits['CHI2'] = top_hits['CHI2'].round(2)
print('=== Top 20 GWAS hits ===')
print(top_hits.to_string(index=False))text
=== Top 20 GWAS hits ===
CHROM POS ID REF ALT CASE_AF CTRL_AF P CHI2
22 23241511 None G A 0.837 0.127 1.22e-293 1348.91
22 23241507 None T C 0.837 0.146 1.37e-264 1215.14
22 23241518 None C T 0.758 0.105 5.45e-263 1207.77
22 21508002 None C T 0.966 0.214 3.75e-254 1167.07
22 21538962 None C T 0.727 0.103 4.24e-233 1070.11
22 21538976 None G C 0.727 0.103 7.91e-233 1068.87
22 20768296 None A G 0.485 0.047 1.17e-223 1026.64
22 25241633 None G C 0.327 0.010 1.95e-223 1025.61
22 20767103 None C T 0.487 0.048 1.71e-222 1021.28
22 20768702 None C G 0.485 0.047 2.45e-222 1020.56
22 23709950 None T C 0.015 0.569 3.15e-221 1015.45
22 23712520 None A G 0.192 0.788 2.65e-217 997.37
22 21475259 None G T 0.696 0.119 4.89e-214 982.33
22 23241523 None G T 0.895 0.261 1.09e-210 966.91
22 20262166 None G A 0.440 0.046 1.66e-205 943.05
22 17824585 None C T 0.305 0.011 3.04e-202 928.02
22 17837775 None C T 0.295 0.008 6.24e-201 921.98
22 17839846 None C G 0.273 0.006 1.67e-200 920.01
22 17834918 None G C 0.316 0.015 5.64e-199 912.97
22 17824838 None G A 0.346 0.019 5.91e-199 912.882.6 PLINK GWAS 명령어 (참고)
실제 운영 환경에서의 PLINK GWAS 명령어입니다.
bash
# Phenotype 파일 준비 (FID IID phenotype)
# 1 1 2
# 1 2 1
# ...
# Case-control GWAS (logistic regression)
plink2 \
--bfile cohort_chr22_qc \
--pheno phenotype.txt \
--covar covariates.txt \
--covar-name PC1,PC2,PC3,PC4,PC5 \
--glm hide-covar \
--out gwas_result
# Quantitative trait GWAS (linear regression)
plink2 \
--bfile cohort_chr22_qc \
--pheno bmi.txt --pheno-name BMI \
--glm \
--out gwas_bmi
# Population structure (PCA)
plink2 \
--bfile cohort_chr22_qc \
--pca 10 \
--out cohort_pca3. Polygenic Risk Score (PRS) 계산
3.1 PRS 의 원리
PRS 는 수많은 변이의 효과 크기를 가중 합산하여 개인의 유전적 질환 위험도를 하나의 점수로 표현한 값입니다.
$$PRS_i = \sum_{j=1}^{M} \beta_j \cdot g_{ij}$$
- $\beta_j$ : 변이 $j$ 의 효과 크기 (GWAS 또는 PGS Catalog 의 weight)
- $g_{ij}$ : 개인 $i$ 의 변이 $j$ 에서의 alt allele dosage (0/1/2)
- $M$ : 사용된 변이 수
3.2 Weight 테이블 구성
실제로는 PGS Catalog, UK Biobank PRS, 연구논문 등에서 weight 를 가져옵니다. 여기서는 방금 수행한 GWAS 결과에서 유의 변이의 log odds ratio 를 weight 로 사용합니다.
python
# GWAS 유의 변이 기반 weight 테이블 생성 (simulation)
# beta 추정: log(case_af/(1-case_af)) - log(ctrl_af/(1-ctrl_af))
# (실제로는 PGS Catalog 의 검증된 weight 사용)
def safe_logit(p, eps=1e-6):
p = np.clip(p, eps, 1 - eps)
return np.log(p / (1 - p))
weight_df = gwas_df[gwas_df['P'] < 1e-5].copy()
weight_df['beta'] = safe_logit(weight_df['CASE_AF']) - safe_logit(weight_df['CTRL_AF'])
# LD clumping 대체: 200kb 윈도우당 top SNP 만 유지 (간략화)
weight_df = weight_df.sort_values('P').reset_index(drop=True)
kept = []
taken_positions = []
window = 200_000
for _, row in weight_df.iterrows():
if all(abs(row['POS'] - p) > window for p in taken_positions):
kept.append(row)
taken_positions.append(row['POS'])
weight_final = pd.DataFrame(kept)[['CHROM', 'POS', 'ID', 'REF', 'ALT', 'beta', 'P']]
print(f'PRS weight 테이블 생성: {len(weight_final):,} SNP')
print('\n=== Weight 테이블 (상위 10) ===')
print(weight_final.head(10).to_string(index=False))
# 파일로 저장 (PGS Catalog 유사 형식)
weight_path = f'{COHORT_DIR}/prs_weights.txt'
weight_final.to_csv(weight_path, sep='\t', index=False)
print(f'\nWeight 저장: {weight_path}')text
PRS weight 테이블 생성: 39 SNP
=== Weight 테이블 (상위 10) ===
CHROM POS ID REF ALT beta P
22 23241511 None G A 3.561328 1.222444e-293
22 21508002 None C T 4.654836 3.751611e-254
22 20768296 None A G 2.951198 1.168717e-223
22 25241633 None G C 3.869458 1.953038e-223
22 23709950 None T C -4.467520 3.152605e-221
22 20262166 None G A 2.788111 1.660187e-205
22 17824585 None C T 3.718971 3.044983e-202
22 18057936 None A G 3.210530 2.557879e-193
22 18947954 None C T -2.342142 8.253241e-188
22 23480925 None G A 2.352517 5.844002e-179
Weight 저장: /tier4/DSC/jheepark/bbp-wgs-data/cohort/prs_weights.txt3.3 개인별 PRS 계산
각 샘플의 genotype 에 weight 를 곱해 합산합니다.
python
# PRS 계산
# (CHROM, POS, REF, ALT) 로 효율적 lookup 을 위한 딕셔너리
weight_dict = {(r['CHROM'], r['POS'], r['REF'], r['ALT']): r['beta']
for _, r in weight_final.iterrows()}
# 샘플 별 PRS 초기화
vcf = VCF(cohort_vcf)
vcf_samples = vcf.samples
prs = np.zeros(len(vcf_samples))
n_used_snps = 0
for v in vcf:
key = (v.CHROM, v.POS, v.REF, v.ALT[0] if v.ALT else '.')
if key not in weight_dict:
continue
beta = weight_dict[key]
# dosage 계산
gts = v.genotypes
dosage = np.array([
(a1 == 1) + (a2 == 1) if a1 >= 0 and a2 >= 0 else 0
for a1, a2, _ in gts
])
prs += beta * dosage
n_used_snps += 1
vcf.close()
print(f'PRS 계산 완료: {n_used_snps:,} SNP 사용')
# 결과 DataFrame
prs_df = pd.DataFrame({'sample': vcf_samples, 'PRS_raw': prs})
prs_df = prs_df.merge(cohort_info[['sample', 'super_pop', 'pop']], on='sample')
prs_df['PRS_z'] = (prs_df['PRS_raw'] - prs_df['PRS_raw'].mean()) / prs_df['PRS_raw'].std()
print('\n=== PRS 기초 통계 ===')
print(prs_df['PRS_raw'].describe())text
PRS 계산 완료: 39 SNP 사용
=== PRS 기초 통계 ===
count 2495.000000
mean 19.722076
std 34.108942
min -23.451407
25% -4.488603
50% 5.912192
75% 30.128629
max 122.128304
Name: PRS_raw, dtype: float643.4 PRS 분포 시각화 (Super Population 별)
python
# PRS 분포 시각화
fig, axes = plt.subplots(2, 2, figsize=(16, 10))
sp_order = ['AFR', 'AMR', 'EAS', 'EUR', 'SAS']
sp_colors = {'AFR': '#e74c3c', 'AMR': '#f39c12', 'EAS': '#2ecc71',
'EUR': '#3498db', 'SAS': '#9b59b6'}
# 1. 전체 PRS 히스토그램
axes[0, 0].hist(prs_df['PRS_raw'], bins=40, color='#3498db',
edgecolor='black', linewidth=0.3, alpha=0.8)
axes[0, 0].axvline(prs_df['PRS_raw'].mean(), color='red', linestyle='--',
label=f'Mean: {prs_df["PRS_raw"].mean():.2f}')
axes[0, 0].set_title('PRS 전체 분포', fontsize=13, fontweight='bold')
axes[0, 0].set_xlabel('PRS (raw)')
axes[0, 0].set_ylabel('샘플 수')
axes[0, 0].legend()
# 2. Super Population 별 PRS boxplot
data_by_sp = [prs_df[prs_df['super_pop'] == sp]['PRS_raw'].values for sp in sp_order]
bp = axes[0, 1].boxplot(data_by_sp, labels=sp_order, patch_artist=True,
medianprops={'color': 'black', 'linewidth': 2})
for patch, sp in zip(bp['boxes'], sp_order):
patch.set_facecolor(sp_colors[sp])
axes[0, 1].set_title('Super Population 별 PRS 분포', fontsize=13, fontweight='bold')
axes[0, 1].set_ylabel('PRS (raw)')
# 3. PRS density by super population
for sp in sp_order:
data = prs_df[prs_df['super_pop'] == sp]['PRS_raw']
if len(data) > 1:
axes[1, 0].hist(data, bins=25, alpha=0.4, label=sp,
color=sp_colors[sp], density=True)
axes[1, 0].set_title('Super Population별 PRS 밀도 분포', fontsize=13, fontweight='bold')
axes[1, 0].set_xlabel('PRS (raw)')
axes[1, 0].set_ylabel('Density')
axes[1, 0].legend()
# 4. PRS percentile (EAS vs 나머지)
prs_df['percentile'] = prs_df['PRS_raw'].rank(pct=True) * 100
axes[1, 1].scatter(prs_df.index, prs_df['PRS_z'],
c=[sp_colors[sp] for sp in prs_df['super_pop']],
alpha=0.6, s=15)
axes[1, 1].axhline(0, color='black', linestyle='-', linewidth=0.5)
axes[1, 1].axhline(1.96, color='red', linestyle='--', linewidth=0.8,
label='Top 2.5% (z=1.96)')
axes[1, 1].axhline(-1.96, color='red', linestyle='--', linewidth=0.8)
axes[1, 1].set_title('개인별 PRS z-score', fontsize=13, fontweight='bold')
axes[1, 1].set_xlabel('샘플 인덱스')
axes[1, 1].set_ylabel('PRS z-score')
axes[1, 1].legend()
# Super Population 범례
from matplotlib.patches import Patch
legend_elements = [Patch(facecolor=sp_colors[sp], label=sp) for sp in sp_order]
axes[1, 1].legend(handles=legend_elements + [axes[1, 1].lines[-1]],
loc='upper right', fontsize=9)
plt.tight_layout()
plt.show()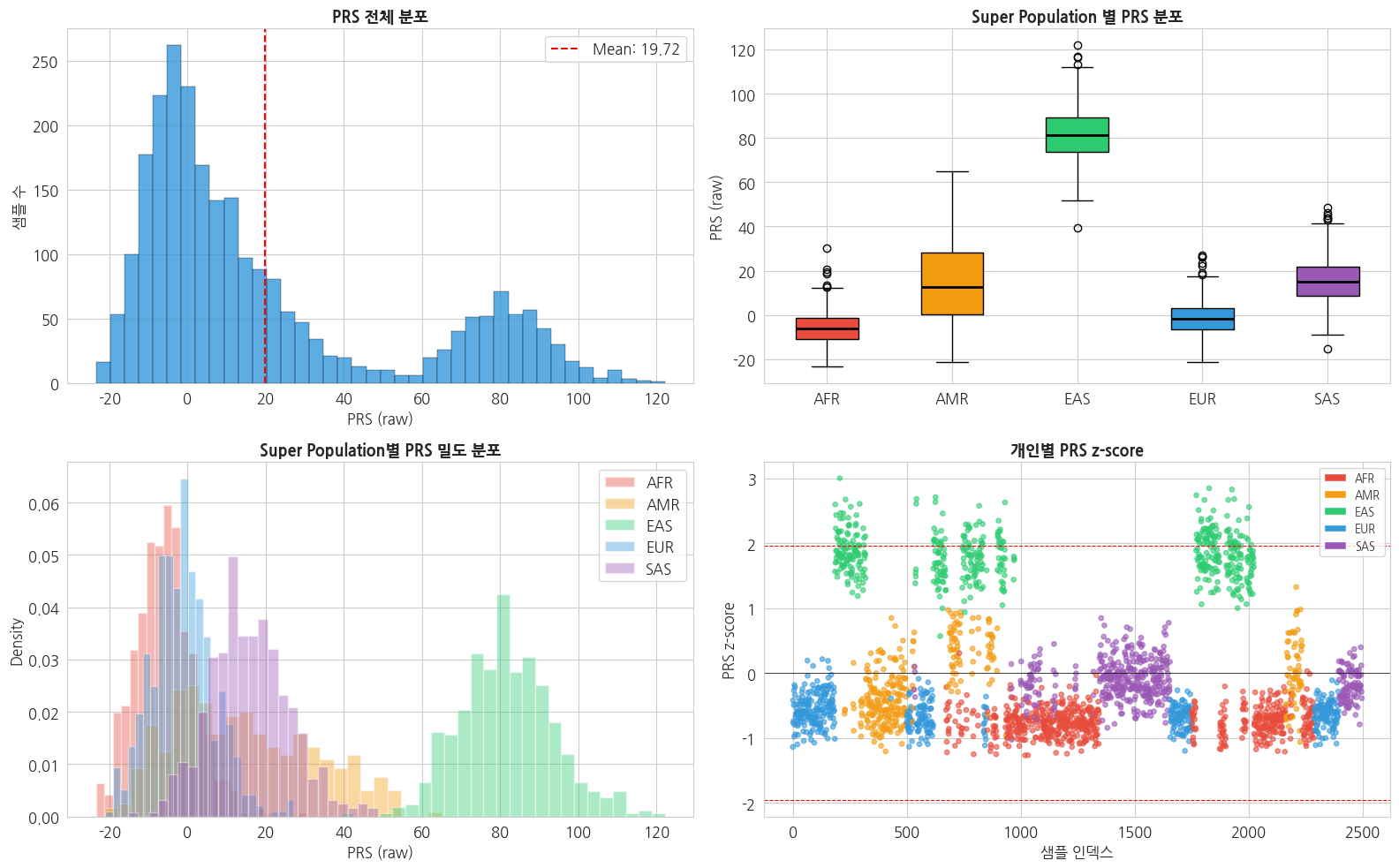
3.5 High-risk / Low-risk 개인 식별
python
# 상/하위 2.5% 개인 식별
prs_df_sorted = prs_df.sort_values('PRS_raw', ascending=False).reset_index(drop=True)
n = len(prs_df_sorted)
high_risk = prs_df_sorted.head(max(1, n // 40)) # top 2.5%
low_risk = prs_df_sorted.tail(max(1, n // 40)) # bottom 2.5%
print(f'=== High Risk (top 2.5%, n={len(high_risk)}) ===')
print(high_risk[['sample', 'super_pop', 'pop', 'PRS_raw', 'PRS_z']].to_string(index=False))
print(f'\n=== Low Risk (bottom 2.5%, n={len(low_risk)}) ===')
print(low_risk[['sample', 'super_pop', 'pop', 'PRS_raw', 'PRS_z']].to_string(index=False))
# Super Population별 high/low risk 비율
print('\n=== Super Population 별 High Risk 비율 ===')
sp_total = prs_df['super_pop'].value_counts()
sp_high = high_risk['super_pop'].value_counts()
for sp in sp_order:
total = sp_total.get(sp, 0)
hi = sp_high.get(sp, 0)
pct = hi/total*100 if total else 0
print(f' {sp}: {hi}/{total} ({pct:.1f}%) in top 2.5%')text
=== High Risk (top 2.5%, n=62) ===
sample super_pop pop PRS_raw PRS_z
HG00458 EAS CHS 122.128304 3.002328
NA18609 EAS CHB 116.879338 2.848440
NA18966 EAS JPT 116.197072 2.828437
NA18526 EAS CHB 113.180469 2.739997
HG01805 EAS CDX 112.143308 2.709589
HG01600 EAS KHV 111.265269 2.683847
NA18976 EAS JPT 111.180711 2.681368
HG00557 EAS CHS 110.987562 2.675706
NA18948 EAS JPT 110.738956 2.668417
NA18567 EAS CHB 110.720712 2.667882
HG01804 EAS CDX 109.594289 2.634858
HG02409 EAS CDX 109.448706 2.630590
HG01599 EAS KHV 108.741445 2.609854
HG00708 EAS CHS 108.669875 2.607756
HG00404 EAS CHS 108.328097 2.597736
HG02152 EAS CDX 107.611050 2.576714
HG00619 EAS CHS 107.130217 2.562617
NA19067 EAS JPT 107.007242 2.559011
NA18538 EAS CHB 105.397192 2.511808
NA18628 EAS CHB 105.189333 2.505714
HG00472 EAS CHS 103.905743 2.468082
HG00533 EAS CHS 103.678875 2.461431
HG01861 EAS KHV 103.615829 2.459582
HG00410 EAS CHS 102.798699 2.435626
HG00674 EAS CHS 102.766316 2.434676
NA18943 EAS JPT 102.349385 2.422453
HG00593 EAS CHS 101.357204 2.393364
NA18615 EAS CHB 101.221127 2.389375
HG00531 EAS CHS 101.210267 2.389057
NA19058 EAS JPT 101.024943 2.383623
HG00464 EAS CHS 100.966370 2.381906
NA18552 EAS CHB 100.796131 2.376915
NA18603 EAS CHB 100.285229 2.361936
NA18997 EAS JPT 100.046046 2.354924
HG02364 EAS CDX 99.794835 2.347559
HG02153 EAS CDX 99.321865 2.333693
NA18550 EAS CHB 99.316636 2.333539
HG00671 EAS CHS 98.202870 2.300886
HG02186 EAS CDX 98.064642 2.296834
HG02049 EAS KHV 97.832711 2.290034
HG00599 EAS CHS 97.711619 2.286484
HG02067 EAS KHV 97.672264 2.285330
HG02134 EAS KHV 97.530037 2.281160
NA18637 EAS CHB 97.509839 2.280568
HG00598 EAS CHS 97.311356 2.274749
NA18623 EAS CHB 97.275798 2.273706
HG00596 EAS CHS 97.275424 2.273695
HG00662 EAS CHS 97.239144 2.272632
NA18625 EAS CHB 96.708300 2.257069
HG02073 EAS KHV 96.346041 2.246448
HG02078 EAS KHV 95.989212 2.235987
NA18613 EAS CHB 95.894468 2.233209
HG00851 EAS CDX 95.857104 2.232113
HG01798 EAS CDX 95.749762 2.228966
HG00407 EAS CHS 95.747654 2.228905
NA18582 EAS CHB 95.742909 2.228765
NA18565 EAS CHB 95.655554 2.226204
NA18641 EAS CHB 95.493544 2.221455
NA18951 EAS JPT 95.320085 2.216369
HG02380 EAS CDX 95.241463 2.214064
NA18602 EAS CHB 95.211779 2.213194
NA18561 EAS CHB 94.760487 2.199963
=== Low Risk (bottom 2.5%, n=62) ===
sample super_pop pop PRS_raw PRS_z
HG03069 AFR MSL -16.498231 -1.061901
HG03172 AFR ESN -16.565194 -1.063864
HG03295 AFR ESN -16.682950 -1.067316
HG02722 AFR GWD -16.701492 -1.067860
HG00257 EUR GBR -16.802519 -1.070822
NA18910 AFR YRI -16.978297 -1.075975
NA18870 AFR YRI -17.121945 -1.080187
HG02941 AFR ESN -17.164695 -1.081440
NA18508 AFR YRI -17.203154 -1.082567
HG03556 AFR MSL -17.239632 -1.083637
NA19922 AFR ASW -17.250699 -1.083961
HG03366 AFR ESN -17.272245 -1.084593
NA20814 EUR TSI -17.287637 -1.085044
NA19257 AFR YRI -17.305514 -1.085568
NA19320 AFR LWK -17.305514 -1.085568
HG03577 AFR MSL -17.389210 -1.088022
HG02420 AFR ACB -17.400785 -1.088362
HG01444 AMR CLM -17.401788 -1.088391
HG03294 AFR ESN -17.414021 -1.088750
HG03049 AFR GWD -17.457121 -1.090013
HG01619 EUR IBS -17.556096 -1.092915
HG01747 EUR IBS -17.601354 -1.094242
NA20530 EUR TSI -17.666297 -1.096146
HG00118 EUR GBR -17.677842 -1.096484
NA19118 AFR YRI -17.693390 -1.096940
NA18507 AFR YRI -17.752867 -1.098684
HG03081 AFR MSL -17.827068 -1.100859
HG02283 AFR ACB -17.904082 -1.103117
NA18881 AFR YRI -18.018774 -1.106480
NA20508 EUR TSI -18.099260 -1.108839
HG02230 EUR IBS -18.197556 -1.111721
NA18915 AFR YRI -18.275029 -1.113992
NA19184 AFR YRI -18.281927 -1.114195
HG01956 AFR ACB -18.379708 -1.117061
HG01174 AMR PUR -18.429985 -1.118535
HG02585 AFR GWD -18.516091 -1.121060
NA19982 AFR ASW -18.577151 -1.122850
NA19247 AFR YRI -18.611958 -1.123870
HG03518 AFR ESN -18.645334 -1.124849
HG02464 AFR GWD -18.718134 -1.126983
NA18933 AFR YRI -18.782278 -1.128864
HG01515 EUR IBS -18.915604 -1.132773
HG03175 AFR ESN -19.032050 -1.136187
HG02339 AFR ACB -19.039326 -1.136400
HG00096 EUR GBR -19.062877 -1.137090
HG02284 AFR ACB -19.266273 -1.143054
NA18853 AFR YRI -19.966319 -1.163577
NA19113 AFR YRI -20.273317 -1.172578
NA18867 AFR YRI -20.273317 -1.172578
NA12716 EUR CEU -21.094669 -1.196658
NA19401 AFR LWK -21.189690 -1.199444
HG03311 AFR ESN -21.190885 -1.199479
HG02281 AFR ACB -21.311308 -1.203010
HG01253 AMR CLM -21.361095 -1.204469
NA19472 AFR LWK -21.546856 -1.209915
HG01885 AFR ACB -21.850131 -1.218807
HG03025 AFR GWD -22.106502 -1.226323
HG03120 AFR ESN -22.590819 -1.240522
HG02334 AFR ACB -22.823956 -1.247357
HG02111 AFR ACB -22.852505 -1.248194
HG02667 AFR GWD -23.361455 -1.263115
HG02645 AFR GWD -23.451407 -1.265753
=== Super Population 별 High Risk 비율 ===
AFR: 0/658 (0.0%) in top 2.5%
AMR: 0/347 (0.0%) in top 2.5%
EAS: 62/503 (12.3%) in top 2.5%
EUR: 0/499 (0.0%) in top 2.5%
SAS: 0/488 (0.0%) in top 2.5%3.6 PRS 결과 저장 및 PRSice-2 명령어 (참고)
python
# PRS 결과 저장
prs_out = f'{COHORT_DIR}/cohort_prs.tsv'
prs_df.to_csv(prs_out, sep='\t', index=False)
print(f'PRS 결과 저장: {prs_out}')
print(f' - 샘플 수: {len(prs_df)}')
print(f' - 컬럼: {list(prs_df.columns)}')text
PRS 결과 저장: /tier4/DSC/jheepark/bbp-wgs-data/cohort/cohort_prs.tsv
- 샘플 수: 2495
- 컬럼: ['sample', 'PRS_raw', 'super_pop', 'pop', 'PRS_z', 'percentile']PRSice-2 명령어 (참고)
실제 환경에서는 PRSice-2 로 다양한 p-value 임계값에서 PRS 를 계산하고 최적 모델을 선택합니다.
bash
# PRSice-2 실행 (GWAS summary stats + target genotype)
Rscript PRSice.R \
--prsice ./PRSice_linux \
--base gwas_summary_stats.txt \
--target cohort_chr22_qc \
--snp SNP --chr CHR --bp BP \
--A1 A1 --A2 A2 \
--stat BETA --pvalue P \
--pheno phenotype.txt \
--cov covariates.txt \
--bar-levels 0.001,0.05,0.1,0.2,0.3,0.4,0.5,1 \
--binary-target T \
--out prs_result결과 해석:
- 최적 p-value threshold : PRSice-2 가 target phenotype 와 가장 잘 맞는 threshold 를 자동 선택
- PRS 설명력 (Nagelkerke R²) : target phenotype 분산의 몇 % 를 설명하는가
- AUC : case/control 분류 성능
4. 요약 및 다음 강의 안내
이번 강의 요약
| 항목 | 내용 |
|---|---|
| 코호트 구성 | Unrelated 샘플만 선별, biallelic SNP, MAF≥0.01 |
| QC 지표 | MAF, Missingness, HWE Chi-square |
| GWAS | 2×3 분할표 + Chi-square, Manhattan/QQ plot, λ |
| PRS | GWAS 기반 weight × dosage 합산, population 간 비교 |
| 산출물 | cohort_chr22.vcf.gz, prs_weights.txt, cohort_prs.tsv |
핵심 포인트
- 코호트 QC 는 GWAS 성패를 좌우 — MAF/Missing/HWE 는 기본, PCA 로 인구구조 반드시 보정
- Manhattan plot + QQ plot 동시 확인 이 GWAS 해석의 기본
- Genomic Inflation Factor (λ) 이 1.1 을 넘으면 원인 파악 후 재분석 필요
- PRS 는 대형 GWAS 의 weight 에 의존 — 원칙적으로 PGS Catalog 등 검증된 weight 사용
- PRS는 인구집단 간 이식성(portability)이 낮아 동일 조상 배경의 weight 사용이 권장됨
주요 도구 명령어 정리
bash
# QC
bcftools view -v snps -m2 -M2 -q 0.01:minor -e 'F_MISSING>0.05' input.vcf.gz
# PLINK QC + GWAS
plink2 --vcf cohort.vcf.gz --make-bed --out cohort
plink2 --bfile cohort --maf 0.01 --geno 0.05 --hwe 1e-6 --make-bed --out cohort_qc
plink2 --bfile cohort_qc --pheno pheno.txt --glm --out gwas
# PCA (인구구조 보정)
plink2 --bfile cohort_qc --pca 10 --out pca
# PRSice-2 (PRS)
Rscript PRSice.R --base gwas_summary.txt --target cohort_qc \
--stat BETA --pvalue P --out prs다음 강의 예고
5강: AI 기반 질병 예측 분석
- 유전체 변이 정보를 feature matrix로 변환
- Mutational signature 분석 및 NMF 기반 패턴 추출
- 머신러닝 모델(scikit-learn)을 이용한 질병 예측 분석